撰文丨Q.Y
責編丨迦漵
急性淋巴細胞白血病(Acute lymphoblastic leukemia , ALL)是一種常見的惡性腫瘤,主要起源於B系或T系淋巴祖細胞,白血病細胞在骨髓內異常增生和聚集並抑製正常造血,導致貧血、血小板減少和中性粒細胞減少;白血病細胞也可侵犯髓外組織,如腦膜、性腺、胸腺、肝、脾,或淋巴結、骨組織等,引起相應病變。由於基礎研究的進展,目前依據ALL不同的生物學特性制定相應的治療方案已取得較好療效,大約80%的兒童和30%的成人能夠獲得長期無病生存,並且有治癒的可能。
作為第一個用於治療急性淋巴細胞白血病的藥物,從五十年前用於治療ALL開始,糖皮質激素(glucocorticoid,GC)一直是治療淋巴系統腫瘤,尤其是急性淋巴細胞白血病的主要藥物。然而,患者對 GC 產生耐葯則是目前ALL臨床治療中常見的難題,與患者的預後直接關聯。常見的耐葯機制有糖皮質激素受體 (glucocorticoid receptor,GR)調控通路的異常等【1】。 儘管進行了大量的研究,糖皮質激素抵抗的機制仍未完全了解。近年來,隨著各種高通量測序技術的興起,以及相應分析方法的出現,多組學、多層次的系統地理解腫瘤的發生發展以及耐葯機制,極大地促進了相關藥物的開發,以及治療手段的改善。
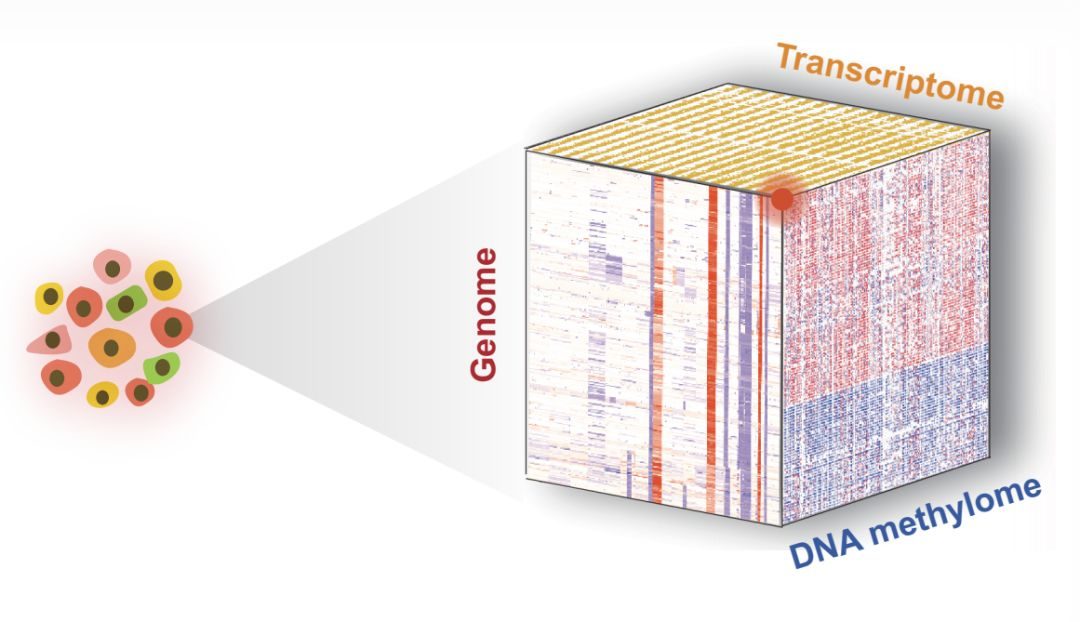
近日,來自澳大利亞的Richard B. Lock研究團隊,在Cancer Cell雜誌發表了Lymphocyte-Specific Chromatin Accessibility Pre-determines Glucocorticoid Resistance in Acute Lymphoblastic Leukemia(https://doi.org/10.1016/j.ccell.2018.11.002)的文章【2】,整合了DNase-seq,ChIP-seq,ATAC-Seq以及甲基化測序技術,從表觀遺傳的角度,闡述了ALL對於糖皮質激素耐葯的的可能原因。


另外一篇同期刊發在Cancer Cell的文章Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia(https://doi.org/10.1016/j.ccell.2018.11.001),也通過整合CRISPR-screening 以及ChIP-seq,ATAC-Seq等多組學手段,找到了對於CLL(慢性淋巴細胞白血病,Chronic lymphoblastic leukemia , ALL)細胞增殖至關重要的轉錄因子PAX5,同時證明了BET bromodomain的抑製可以破壞CLL core regulatory circuitry從而發揮較強的抗腫瘤活性【3】。


糖皮質激素在多年前就已經被證明,能夠通過結合糖皮質激素受體(NR3C1,Glucocorticoid Receptor,GR),並激活GR導致下遊基因轉錄水準的改變,特異性地減弱ALL細胞的增殖能力並誘導ALL細胞的凋亡,同時不影響其他組織細胞的存活【4】。 作者基於這一理論基礎,提出了糖皮質激素誘導的細胞死亡是由一些被糖皮質激素受體所調控的淋巴細胞特異性的基因調控元件所介導的假設。
基於這一假設,作者首先通過使用公共的資料庫資源,收集了82種不同類型的細胞系的DNase-seq 數據,繪製了淋巴細胞和大量的非淋巴細胞類型中的DNase I-hypersensitive sites (DHSs)圖譜,鑒定出超過11,000個淋巴細胞特異性的染色質開放區域(lymphocyte-specific open chromatin structure,LSO),這些區域大部分都落在了基因或者基因的啟動子區域。
為了研究GC在淋巴細胞中的作用是否與這個特異性的LSOs相關,作者使用ChIP-seq的方法,鑒定GR是否能夠與LSO結合,ChIP-seq結果顯示,在GC的刺激下,GR與LSOs的結合能夠顯著增強,與非淋巴細胞特異性的染色質開放區域的結合併無顯著差異,提示了LSOs可能被GC所調節,也就是說,GC特異性地在淋巴細胞中發揮作用可能是通過LSOs所介導的。
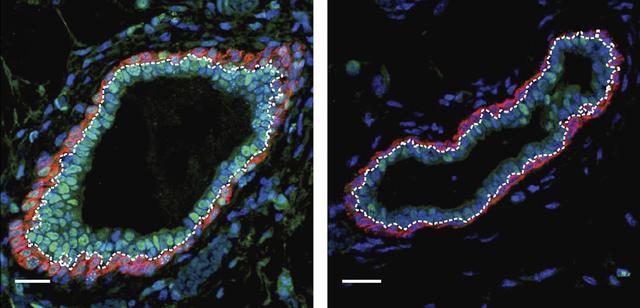
為了確定糖皮質激素抗性ALL細胞中可能失調的位點,作者關注於糖皮質激素敏感以及抵抗的細胞中差異表達的,並且位於糖皮質激素受體結合位點附近的可能被GC所調控的基因。作者通過系統地整合RNA-seq,GR以及H3K4Me3,H3K27Ac的ChIP-seq,以及ATAC-Seq的數據,作者找到了近400個candidate基因,其中198個基因表達水準上調,191個表達水準下調。此外,通過DNA甲基化測序以及ATAC-Seq,鑒定出了42個candidate LSOs,這些LSOs在糖皮質激素抗性白血病細胞中存在高度的DNA甲基化,同時ATAC-seq分析顯示這部分染色質在糖皮質激素耐受的細胞中處於Closed的狀態,Chromatin marker也顯示此處處於不活躍轉錄的狀態。這一結果在更多的敏感以及糖皮質激素抵抗的ALL的PDX模型中得到了進一步的驗證。
綜上,作者首先從0.82million DHS peaks中,找到了超過11000個LSOs,進一步證實大約10%(1177)的LSOs能夠被GR所識別並結合。作者將這些數據與RNA-seq,ChIP-seq,DNA甲基化測序,ATAC-Seq測序相整合,進一步篩選出了42個LSOs以及40個相關的基因。接下來作者將對這些候選基因以及LSOs做進一步研究,以探究其作用以及作為潛在的治療靶點的價值。
值得注意的是,在42個抗性淋巴細胞中失調的糖皮質激素結合的LSOs中,有一個對應於BCL2L11基因的intronic glucocorticoid receptor-binding region(IGR)位點。進一步的實驗表明, IGR在糖皮質激素的刺激下,GR以及CTCF與之結合增加,染色質可及性和組蛋白乙醯化都有增加,這種增加在GC耐葯的細胞中顯著降低,作者進一步使用Luciferase實驗證實了IGR具有增強轉錄的活性。有趣的是,作者發現GR以及CTCF雖然都與IGR有較強的的結合,但是結合的位點卻有差別,存在一定的互斥的現象。提示了CTCF可能也在IGR對於基因轉錄的調節中起著至關重要的作用。進一步的研究發現,CTCF在糖皮質激素處理的敏感細胞中對BCL2L11 IGR的結合顯著增加,糖皮質激素耐葯的細胞不管在basal條件下還是在糖皮質激素刺激下,CTCF的結合較敏感的細胞都顯著降低。
遠端增強子可以通過與Cohesin複合物以及CCCTC結合因子CTCF形成Chromatin Loop與近端啟動子相互作用並控制其活性,結合位點的突變與癌症具有較強的相關性【5-7】。作者進一步使用在染色體構象捕獲(3C)方法驗證在這一區域的染色質構象。實驗結果表明,在敏感細胞中,GC處理能夠顯著提高IGR與Promoter的Looping, 在不敏感的細胞中這一Loop結構則檢測不到,這與糖皮質激素不能誘導BCL2L11轉錄上調的結果是一致的。另外,在糖皮質激素敏感細胞中敲除IGR這一元件後,糖皮質激素誘導的BCL2L11轉錄激活以及細胞死亡現象就會消失。充分說明了這一區域對於糖皮質激素的敏感性起著至關重要的作用。


染色質可及性通過pioneering transcription factors建立,其可以與核小體DNA相互作用並介導染色體重構複合物的募集以產生可以被轉錄因子結合的無核小體區域【8】。儘管負責淋巴細胞特異性的的染色質可及性和BCL2L11 IGR區域具有調控轉錄活性的具體機制仍有待完全闡明,但PU.1(一種具有先驅活性的轉錄因子),能夠與IGR區域結合,並且在糖皮質激素抵抗的的ALL細胞中結合顯著降低。此外,IGR中的DNA甲基化在糖皮質激素抗性ALL中顯著增加,提示了DNA甲基化在染色質構型中的作用。與此假設一致的是,用5-azacitidine(DNA去甲基化劑)處理糖皮質激素抗性ALL細胞,逆轉該位點的DNA甲基化,能夠增強糖皮質激素誘導的BCL2L11上調,並增強體外糖皮質激素反應。另外,在體內使用decitabine(一種臨床DNA去甲基化藥物)與糖皮質激素聯合治療,GC治療活性也增加。
儘管關於糖皮質激素抵抗的表觀遺傳機制仍有許多尚待了解,但本研究提示了表觀遺傳學介導的BCL2L11 IGR沉默的潛在作用。揭示了GC耐葯的可能原因:在耐葯的細胞中,由於IGR區域的高度甲基化導致PU.1對其的結合減弱,從而導致不能後續地被GR以及CTCF結合形成Loop結構,從而抑製了相關基因的轉錄,導致了耐葯的發生。此外,染色質可及性的變化也可能與DNA甲基化有關,並且該位點的先驅轉錄因子的作用仍有待充分闡述。


早在2016年,William J. Greenleaf與 Monte M. Winslow合作在Cell雜誌發表文章【9】,在 SCLC小鼠模型中分離出原發腫瘤和轉移灶的癌細胞,使用ATAC-Seq技術研究癌症轉移擴散的機制。試圖從基因組的染色質可接近性的角度出發找到相關的分子機制,找到了在轉移的細胞中,基因組許多的遠端調控元件處於更加open的狀態。同時轉錄因子Nfib高度富集在這些位點,進一步的實驗有力地證明了Nfib能夠促進腫瘤的轉移。並驅動了SCLC細胞的轉移能力。
今年早些時候中國科學院上海藥物研究所耿美玉、丁健和譚敏佳聯合研究團隊在Cell上發表了題為Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors的研究成果【10】,揭示了EZH2抑製劑對大部分實體瘤治療無效的分子機制並提供了可能的協同抑製表觀遺傳互動調控解決方案,並對當前腫瘤的靶向治療提出了示範性方案,對於當前EZH2抑製劑臨床研究的困境破局具有重要的指導意義。Bioart 。 另外,今年初,美國丹娜法伯癌症研究院(Dana-Farber Cancer Institute)和Harvard-MIT Broad研究所等研究機構聯合發表題為Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma和A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing兩篇文章【11, 12】。兩個研究小組從不同角度出發,一致發現一組被稱為「染色質重塑複合物(chromatin remodeling complex,SWI/SNF)」的DNA包裝壓縮調節蛋白,通過自身變化嚴格控制免疫檢查點阻斷。ARID2、PBRM1和 BRD7等基因參與複合物不同組件編碼。SWI/SNF可打開緊密纏繞在一起的DNA,使特定基因能被讀取、激活和轉錄。


因此,隨著測序技術的發展,以及三維基因組學的興起(北大李程組綜述:三維基因組學及在疾病中的應用),對於腫瘤發生發展以及腫瘤耐葯機制有了更深入的認識,這些基礎研究的突破,有望帶來更特異、更有效的、更精準的抗癌靶向治療手段。
參考文獻
1. Paugh, S.W., et al., NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nature genetics, 2015. 47(6): p. 607.
2. Jing, D., et al., Lymphocyte-Specific Chromatin Accessibility Pre-determines Glucocorticoid Resistance in Acute Lymphoblastic Leukemia. Cancer Cell, 2018. 34(6): p. 906-921.e8.
3. Ott, C.J., et al., Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer cell, 2018.
4. Wyllie, A.H., Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature, 1980. 284(5756): p. 555.
5. Watrin, E., F.J. Kaiser, and K.S. Wendt, Gene regulation and chromatin organization: relevance of cohesin mutations to human disease. Current opinion in genetics & development, 2016. 37: p. 59-66.
6. Busslinger, G.A., et al., Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature, 2017. 544(7651): p. 503.
7. Ong, C.-T. and V.G. Corces, CTCF: an architectural protein bridging genome topology and function. Nature reviews Genetics, 2014. 15(4): p. 234.
8. Zaret, K.S. and S.E. Mango, Pioneer transcription factors, chromatin dynamics, and cell fate control. , 2016. 37: p. 76-81.
9. Denny, S.K., et al., Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell, 2016. 166(2): p. 328-342.
10. Huang, X., et al., Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell, 2018. 175(1): p. 186-199.e19.
11. Miao, D., et al., Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science, 2018. 359(6377): p. 801-806.
12. Pan, D., et al., A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science, 2018. 359(6377): p. 770-775.
BioArt,一心關注生命科學,只為分享更多有種、有趣、有料的資訊。關注投稿、合作、轉載授權事宜請聯繫微信ID:fullbellies 或郵箱:[email protected]