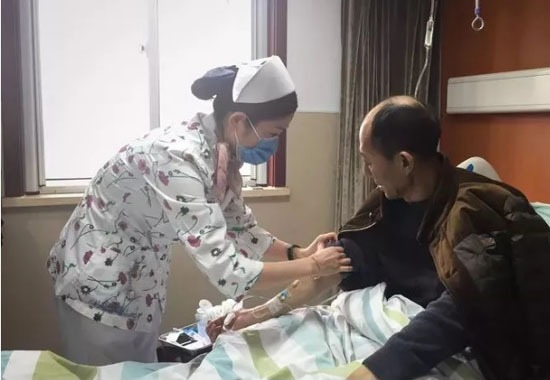
2018年11月26日,yoyo(化名)迎來了她8歲的生日。母親秦可佳早早就下班回家,給她買了生日蛋糕和一頂皇冠。戴著皇冠的yoyo四處跑動,一邊拍手一邊叫喊著,看起來十分高興。
早在一歲半的時候,她能叫爸爸、媽媽、奶奶,可如今她只能“咿啊”地叫著,不知面前的蛋糕為何物。為了不讓女兒打翻蛋糕,秦可佳從後面抱住了yoyo,母女倆一起拍了張照。

yoyo5歲生日 受訪者供圖
秦可佳從不把女兒生日的照片分享到朋友圈,“從yoyo出生開始,她的生命就在倒計時”。yoyo體內的一個基因出了問題,導致她患上一種罕見病,她的身體、感官和智力都在不斷退化。
為了打破這個“死亡沙漏”,秦可佳多年來奔波於國內外尋醫問藥,將最後的希望寄托於基因治療。但就在生日聚會後,她在新聞上看到,一對名為露露和娜娜的基因編輯嬰兒已經出生。
儘管罕見病基因治療與基因編輯嬰兒有本質不同,但後者引發的巨大爭議還是讓基因治療蒙上一層陰影,也讓努力了五年的秦可佳平添了幾分擔憂。
致命基因
今年38歲的秦可佳與大自己一歲的宋濤結束了十年愛情長跑走到一起,碩士畢業後兩人來到北京投身於航天事業。婚後兩年,女兒yoyo出生了。
剛出生時yoyo就有一頭濃密亮澤的頭髮,臉蛋柔軟嫩白,秦可佳陷入了前所未有的幸福之中。兩歲之前,yoyo和別的孩子一樣,一步步學會翻身、坐爬、站立、走路。只有一樣讓秦可佳感到隱隱擔心,yoyo的語言功能遲遲沒有進步。

1-2歲時的yoyo 受訪者供圖
儘管如此,看到yoyo萌萌的樣子,秦可佳與宋濤並沒有多想,直到發現慢慢長大的yoyo有些異樣——
“她總是到處亂跑,沒有主動叫過誰,對一切事物感到好奇。”聽不到一聲“媽媽”,這讓秦可佳十分著急。
2013年5月,秦可佳和宋濤奔赴靶場執行神舟十號發射任務,期間只和yoyo通過電話聯繫。電話裡yoyo總是笑著,秦可佳感到女兒很想說話,卻不會表達。
等到7月任務結束回到老家,秦可佳進門就想抱抱女兒,yoyo有些冷漠,抱完轉身就走了,仍然沒有主動叫媽媽。
5天后回到北京,夫妻倆直接帶著yoyo去到首都兒科研究所。初步檢查顯示,yoyo的發育商偏低,判斷智力發育遲緩;一周後,核磁共振顯示yoyo的大腦出現了脫髓鞘病變,這是一種常見於阿爾茨海默病(老年癡呆症)的症狀。
秦可佳回憶,當時她就跟瘋了一樣,她不明白女兒怎麽會得這樣的病。夫妻倆跑遍了北京的各大醫院,隻記得一位醫生說,他從來沒見過這病在這麽小的孩子身上出現。
9月的北京驕陽似火,而在中國人民解放軍總醫院(301醫院),秦可佳形容自己的心和冰窖一樣冷。她和宋濤被醫生喊進了辦公室。在此之前,她得知女兒患上的可能是一種叫做黏多糖Ⅲ型溶酶體貯積症(以下簡稱MPSⅢ)的遺傳代謝病。但此時,他們仍然希望能得到不一樣的結論。
然而那一整個下午,秦可佳哭著聽完了醫生的話。
“你們要有心裡準備,孩子會在接下來的幾年裡慢慢失去所有功能,包括記憶、理解、語言、行動甚至吞咽,他們大多活不到青春期……”
301醫院小兒內科副主任醫師孟岩告訴秦可佳,yoyo的病是由一種單基因缺陷引起的罕見病。基因缺陷導致yoyo體內天生缺少一種酶來分解黏多糖分子,導致黏多糖在全身細胞內堆積,隨著時間的推移破壞髒器、骨骼、神經系統……
孟岩形容黏多糖病,“如同一個房間,垃圾堆得越來越多卻無法清理,最終人無法入住。”
其中Ⅲ型直接病變於中樞神經系統,yoyo的大腦會隨著年齡的增長慢慢萎縮,失去所有功能。孟岩說,Ⅲ型黏多糖無藥可治。
而所有黏多糖貯積征的發病率為十萬分之一,根據發病率計算,中國至少有1000人患有這種病。
回去的路上,秦可佳強忍著淚水。yoyo走著跳著,對發生在自己身上的事一無所知。

奔跑中的yoyo 受訪者供圖
“生機”與“絕望”
這不是孟岩第一次接觸“黏寶寶”(黏多糖溶酶體貯積症患兒)。早在7年前,鄭芋帶著女兒飛飛(化名)從西安趕到北京,想為女兒尋找一線生機。
在飛飛4歲半的時候(2001年),鄭芋送她去上舞蹈班,老師反映飛飛的腳常常使不上力氣,直到在醫院拍片後才發現飛飛的骨骼異常,原因不明。
“當時醫生說可能是黏多糖,我還以為是糖尿病之類的毛病。”這是鄭芋第一次聽說這個名詞,走了七八家醫院後,醫生告訴她,飛飛患的是Ⅳ型黏多糖貯積症,“孩子長不高了,一般10多歲就沒了,給孩子吃好喝好吧。”
“哪怕她缺哪個器官我給她,只要能救我女兒的命,但這種病我想給也給不了……”為了給女兒治病,2005年底鄭芋辭去工作,隻身來北京,“我就不信女兒只能等死。”
正如鄭芋所說,並非所有的黏多糖患兒都毫無生機可尋。
2005年10月,一個叫健健的男孩在廣東省第二人民醫院的手術室裡接受了中國首例黏多糖患兒骨髓移植手術。負責這台手術的血液科主任王玲介紹,黏多糖病十分複雜,根據基因的類型以及所編碼酶的不同,至少分為7種大型和16種亞型,每種類型的臨床表現和治療方法都不相同。
當時根據基因檢測和酶學結果判斷,健健屬於Ⅰ型,國內出現較多的是Ⅰ、Ⅱ、Ⅳ型,發病率均為十萬分之一。
健健的手術很成功,移植兩個月後“天門”完全閉合、身高92厘米、肝脾恢復正常,智力發育指數也慢慢升高。
2010年,健健的母親王秀群接受成都商報回訪時說,健健現在活潑調皮,正在上幼稚園。他很愛吃,個子也長得快。最近一次測身高,健健是1.2米,在幼稚園裡算中等偏上。
2006年,當鄭芋得知骨髓移植是唯一的治療手段時,她灰暗的世界裡亮起了一盞燈,“當時第一反應就是把房子賣了,為了救孩子,我什麽都可以不要。”
但這盞燈很快就滅了。
廣東省第二人民醫院血液科主任歐瑞明介紹,從2005年至今,二院僅做過兩例骨髓移植(包括健健),第二例經過基因檢測後發現也是Ⅰ型黏多糖患者。即使到今天,專家認為只有Ⅰ、Ⅱ、Ⅵ型的患者適合做移植,這其中還要看患者局部受損的情況、骨髓提供者的狀態等等,條件十分“苛刻”。
多年後王玲在一篇博文中寫道,骨髓移植的效果也取決於眾多因素。其中之一就是,移植時的年齡最好在18個月以內,趕在過度積累的黏多糖給組織和器官帶來不可逆性損害之前實施。
而對於Ⅳ型患者來說,身體器官和骨骼的變化較慢,不一定適合骨髓移植。前世界最矮男子何平平身高僅74.61厘米,他也是一名Ⅳ型患者,終年21歲。
鄭芋從醫生那裡得到的答案是——飛飛不適合做骨髓移植,失敗的概率很高,而且飛飛的發病過程穩定,不會損害到大腦,即使進行骨髓移植,效果也並不一定理想。
鄭芋猶豫了,她選擇了保守。
和yoyo不同,飛飛知道發生了什麽。9歲那一年,飛飛突然問鄭芋,為什麽我長不高?鄭芋只能安慰她。
女孩都愛美,因為腿腳不便,飛飛上樓梯要麽由爺爺背,要麽自己一步步爬。她覺得自己上樓的姿勢難看,所以每次都讓鄭芋走在前面,不讓她回頭看自己。
在母親鄭芋眼裡,飛飛多數時候是樂觀的,但她也跟奶奶說過類似“白發人送黑發人”這樣的話。
女兒患病後的很長一段時間裡,鄭芋不會笑也不社交,整天埋頭搜索關於黏多糖的資料,到處尋醫,卻發現有些醫生對罕見病知之甚少。
絕望籠罩著她。和秦可佳一樣,面對疾病,兩個母親必須和時間賽跑,卻不知希望在哪。

秦可佳抱著yoyo,此時yoyo6歲。 受訪者供圖
“孤兒藥”困境
2017年10月,中華醫學會兒科學分會血液學在《中國小兒血液與腫瘤雜誌》發表了一篇名為“異基因造血幹細胞移植治療黏多糖貯積症兒科專家共識”的文章。裡面提到,目前MPS的主要治療方法為酶替代治療(ERT)和異基因造血幹細胞移植(HSCT)。
酶替代療法的原理很簡單:人體缺什麽酶,就從外部進行補充。這不僅可以改善部分患者的臨床症狀,同時也降低了併發症。
秦可佳和鄭芋早就了解到這種療法,幾乎所有黏多糖患者的家屬也都知道,可很少有人能夠用上。
歐瑞明解釋說,酶替代的藥物大多在國外研製生產,目前在國內沒有上市。“沒有國家藥監部門的許可證即會被認定為假藥,如果在國內銷售就是違法的,就像電影《我不是藥神》裡的那樣。”
北京協和醫學院遺傳學家黃尚志2013年在接受新京報採訪時分析,從生產角度而言,與常見病相比,罕見病人群相對較小,而投入藥物生產研究過程耗費較高,藥廠回流的錢就會少。因此這些藥被形象地稱為“孤兒藥”。
目前,國內只有極少數“孤兒藥”被個別省市納入了醫保。如果患者自費購買酶替代藥物,每年花費大約在150-200萬人民幣左右,且需終身用藥。
鄭芋身邊僅有兩個家庭在國外買過藥,其中一個孩子的父親是商人,五年花費近千萬。每次買藥直接給美國藥廠匯款,藥廠人員將藥品轉運到香港,購買人再去香港經中間人取藥。
“56萬元人民幣,訂購了40支注射用藥,每支14000元。這是孩子4個多月的用藥量。什麽時候錢沒了,孩子也就沒了。”
儘管多年來醫學專家、學者都不遺余力呼籲將黏多糖等罕見病納入醫保範疇,但截至目前收效甚微。
黃尚志在採訪中說道,“相關部門多將財力投入在多發病、常見病,以及非典、H7N9等對社會穩定有重大影響的疾病上。而諸如黏多糖等相對罕見的病症,關注度不夠。”
秦可佳的困境在於,即使她有這個經濟能力,yoyo也無藥可用——由於Ⅲ型發病集中在中樞神經系統,酶作為大分子被注入人體後無法通過血腦屏障進入大腦,酶替代療法對她“關上了大門”。
yoyo在5、6歲的時候特別多動,喜歡搖晃椅子和門。由於逐漸失去感知外界的能力,她只能通過這種的方式刺激自己,比如摔東西。
“家裡的碗已經換了幾批,電視機換了三個,手機已經數不清幾個了。”秦可佳說,yoyo甚至會把手指咬破,為此她曾經不得已給女兒帶上特製手套。
如今yoyo已經8歲,進入到病症中後期。她不再多動,行動能力和吞咽能力進一步下降。2017年做的24小時腦電監測顯示,她的睡眠波出現嚴重異常。
yoyo經常在半夜裡醒來,秦可佳唯一能做的只是抱著她,直到她疲倦睡去。“不論女兒變成什麽樣,我永遠會給她毫無保留的愛。”但她清楚地意識到,留給女兒的時間不多了。
再過幾年,yoyo就會失去吞咽功能,那時就要在她胃上開一個洞,用導管將流食注入體內;接著腦萎縮會讓她癲癇不斷發作,呼吸能力也會變差,她的生命隨時可能終止。
希望的曙光
秦可佳不上班時,幾乎24小時守著yoyo,防止她摔倒、磕傷。除此以外,她經常一個人在家附近的肯德基查資料、寫文章。也許女兒永遠也無法像餐廳兒童樂園裡的孩子一樣嬉戲玩耍,但她沒有放棄尋找希望。
2014年,她通過美國臨床試驗數據庫(ClinicalTrials.gov)找到了一條關於MPSⅢA的記錄。一家名為Lysogene的法國公司在2014年發布了一項臨床試驗,標題的最後寫道“Gene Therapy”,也就是基因治療。這是秦可佳第一次看到這個詞。
在這份公開記錄中,已經有四名MPSⅢA患者進行了藥物注射,直到今天,臨床試驗仍處於1/2期。
秦可佳立即聯繫到這家公司的CEO,一位46歲的法國媽媽Karen Aiach。Karen說,自己的女兒Ornella也是一位MPSⅢA患者,已經用藥2年多,目前狀態穩定。
此時秦可佳並不明白什麽是基因治療,也不知道試驗是否成功,但她在絕望中看到一絲轉機。至於風險,“我沒得選,哪怕是希望渺茫我也會去找。”當時秦可佳在國內的搜索引擎裡輸入“基因治療”,只有一條相關資訊,除此以外找不到任何報導和文章。
秦可佳說,根據Lysogene公布的數據,一期臨床試驗的安全性已經得到驗證。
而Karen在2015年接受美國財經雜誌《富比士》採訪時說道:Ornella依然是一名MPSⅢA患者,只是從6歲起開始了治療。從過去三年的情況來看,她好轉了許多,不再過度活躍,睡眠也很好。這改變了她的生活,也改變了我們的生活。現在,我有一個會笑的孩子,這是我能想到的最好的結果。

Karen的女兒Ornella
作為一名科研工作者,秦可佳習慣用詳實的數據和資料來分析判斷,基因治療是否可靠。
2016年3月,美國Abeona公司在臨床試驗數據庫發布了一項試驗,對象同樣為MPSⅢA患者,參與人數為16人。
在Abeona公司官網上,可以查詢到更多關於臨床試驗的數據與內容。其原理可以概括為“基因替代”(Gene Replacement),即通過基因載體(通常為改造過的病毒)導入一段有獨立功能的基因進入體細胞,代替已經發生突變的基因進行獨立表達,發揮其原本應該有的功能。
這個案例讓秦可佳眼前一亮,其中一位受試者美國女孩Eliza比yoyo大一歲,也是Ⅲ型,這更讓她感到希望就在眼前。然而由於我國尚未建立罕見病醫保,對方拒絕了yoyo的試藥申請。

Eliza(左二)和她的家人
但此時秦可佳確信自己找到了方向,她把yoyo最後的時間都押在了基因治療上,“我要研製出自己的孤兒藥。”
正如黃尚志此前所說,由於市場太小,鮮有人願意將精力和金錢投資於罕見病藥物研發上,秦可佳只能自己尋找科學家、自己募集資金進行研發。“最困難的是資金,光一隻模型老鼠的費用就高達上萬元,一隻用於試驗的猴子甚至需要30萬元。”
只有當基礎研究、動物試驗等前期準備工作完成,針對yoyo的項目才有機會進行學術和倫理審查。即使通過審查進入到臨床試驗階段,yoyo也只能被稱作“受試者”,對她進行的並非治療,而是試驗,仍然有失敗的可能。
“所有人都覺得我是瘋子,認為我病急亂投醫,但正因為研究過國外公開的資料和案例,它給了我希望和努力的方向。路再難,我也要拚一把。”
正如Eliza的父親格倫·奧尼爾在同意女兒接受試驗之前說的那樣,“這是她唯一的機會。”(It was her only shot)

yoyo和秦可佳,此時yoyo7歲 受訪者供圖
不得已的“賭局”
“我是航天人,基因替代就好比火箭搭載衛星進入太空。火箭是病毒載體,衛星是起作用的正常基因,我們需要什麽樣的基因,就利用病毒包裝好這個基因送進人體細胞。在這個過程中,火箭是運輸工具,用什麽衛星我們說了算。”
秦可佳打了個比方來解釋基因治療的機理,她深信未來這會成為治療罕見病的主攻方向。
哈佛大學醫學研究所成員田禾在此前發表於澎湃新聞的《基因療法解題》一文中解釋道,自然界中有一種善於跨域細胞膜的微生物——病毒。科學家想到,如果將需要添加的基因片段插入病毒的基因組中,也許可以依靠病毒將基因片段帶進活體細胞中。
然而病毒不是細胞,不能獨立分裂,要寄生在宿主細胞內才能自我複製。這種複製是以自身健康為代價的,免疫系統會與病毒展開搏鬥,宿主會出現炎症反應。
所以天然存在的病毒是不能作為運輸載體的,病毒需要經過改造,失去細胞內複製的能力,還要盡可能避免觸發免疫反應,危及宿主生命。
過去20多年來,基因治療的發展經歷了一些波折。
1999年,美國賓夕法尼亞大學的詹姆士·威爾遜開發出一種腺病毒載體(Adenovirus),並開展了一系列臨床試驗。其中一項試驗針對一種罕見的代謝疾病,患者缺少尿素代謝的一種酶,大部分無法活到14歲。少年傑斯·格爾辛基已經與這種病搏鬥了18年。
研究人員把製造酶的基因裝進了被“騰空”的病毒載體裡,約1兆個載體被注入格爾辛基體內,導致他產生了激烈的免疫反應。幾天后,格爾辛基的生命永遠定格在18歲,這是第一個死於基因治療的臨床試驗者。
美國北卡羅萊納大學藥學院教授肖嘯解釋道,格爾辛基因為急性免疫反應而死亡,靜脈注射高劑量腺病毒是威爾遜的致命錯誤,當然還有一系列違規行為。這其中就包括之前在猴子身上進行的類似試驗時,已經導致實驗動物死亡,然而臨床試驗並沒有及時叫停。

格爾辛基
這是基因療法歷史上的一次慘痛教訓,隨後威爾遜被美國FDA禁止十年不許涉及臨床試驗,而這期間大部分基因療法的臨床試驗均以失敗告終。
直到18年後,美國監管部門才批準了一項關於遺傳性視網膜病變的基因治療,並誕生了首款在美國獲得批準的基因治療藥物。根據FDA官網數據,截至目前全球基因治療臨床試驗大約有2000多例,已批準上市藥物有6個,尚無安全性事件報導。
這其中,腺相關病毒(Adeno-associated virus,簡稱AAV)成為了新的“火箭”。肖嘯介紹,和當年所使用的腺病毒不同,AAV是脊椎動物病毒中很少見的非病原性病毒,也是先天性缺陷病毒,本身不能複製,需要在其它致病性病毒存在時才能複製,同時抑製那些病毒。因此可以說是一種“好”病毒。
儘管如此,即使選對了“火箭”,“火箭”的量應該控制在多少等一系列問題仍然需要縝密研究。
田禾說,格爾辛基的悲劇提醒著所有醫學研究者,在試驗新療法時,必須戰戰兢兢、如履薄冰。
繞不開的倫理問題
原國家衛生部發布的《人類輔助生殖技術規範》規定:“禁止以生殖為目的對人類配子、合子和胚胎進行基因操作”;《人胚胎幹細胞研究倫理指導原則》也明確規定,不得將已用於研究的人囊胚植入人或任何其它動物的生殖系統。
2018年年末的基因編輯事件在科學界引起軒然大波。基因編輯技術權威、CRISPR基因編輯專利持有人張鋒此前接受澎湃新聞採訪時表示,多數科學家都認同基因編輯嬰兒的實驗沒有任何意義,而且是不道德的。
在yoyo生日的那天晚上,秦可佳不停地收到來自身邊醫生、朋友的詢問,起初她沒有在意,“這起事件很明顯與罕見病的基因治療是完全不同目的和結果。“
肖嘯也指出,現階段基因治療主要有基因替代和基因編輯兩種方式,不論哪一種均用於治病救人,隻作用於體細胞而非生殖細胞,這與基因編輯嬰兒的實驗有本質不同。
但秦可佳擔心,基因編輯嬰兒事件引起的連鎖反應會使國內正常的基因治療被迫收緊,“如果這樣,等待救命的罕見病患兒就沒有希望了。”

基因治療有望在未來5到10年惠及大眾 人民日報 圖
在這次風波前,我國的基因治療研究已經取得了顯著進展。
中國生物技術發展中心主辦的微信公眾號“新葉社”在《基因治療的國內外研究進展》一文中介紹道,我國基因治療研究及臨床試驗與世界發達國家幾乎同期起步,目前已經有3個基因治療產品上市,此外還有20多個針對惡性腫瘤、心血管疾病、遺傳性疾病的基因治療產品進入了臨床試驗,其中在臨床試驗網上登記的基因治療臨床試驗方案就有70多個。
秦可佳反覆強調,罕見病用藥與常見病不同,“首先是極大的迫切性,其次是患者子樣少。目前已發現的罕見病7000余種,90%左右無藥可治,很多孩子自出生就承受難以想象的痛苦。對他們而言,有藥可用才是最重要的。”
在第二屆國際人類基因組編輯峰會上,倫理學家邱仁宗對眾多媒體強調,不管是針對體細胞進行的基因治療,還是用CRISPR對生殖細胞進行的基因編輯,都應該安全可靠,嚴格按照程式來走。
中國孤兒藥創新聯盟發起人鄭維義介紹,在進行臨床試驗前,找到治療疾病的化合物等基礎研究就需要約3-4年,罕見病藥物臨床試驗需要2-3年,整個周期十分漫長。
秦可佳覺得自己等不起了。yoyo的情況一年比一年糟糕,她期望女兒有機會,哪怕只是維持住現在的狀態,而不是等到她無法進食、只能靠呼吸機生活,藥才遲遲出現。那時候,再救她反而是種殘忍。
“如果我私下找個診所給yoyo用藥,那跟基因編輯嬰兒的實驗性質就沒有區別,整個行業發展會被攪亂,傷及無辜。”秦可佳說,在經歷過那麽多挫折後,她變得更加理性。
“母親的呼籲”
秦可佳從來就不是一個柔弱的女性,這一點鄭芋深有體會。
早在2012年,鄭芋牽頭成立了北京正宇黏多糖罕見病關愛中心,但秦可佳認為僅僅“關愛”還不夠,罕見病更需要的是被社會關注,通過社會力量推動政策和科研進步。
2015年秦可佳建立了cureyoyo網站,幫助MPS患兒家庭提供護理指導和研究資訊;隨後,她牽頭髮起了北京天使媽媽慈善基金會SF罕見病專項基金。2018年,基金向華東理工大學生物工程學院定向捐贈110萬用於MPSⅢA基因治療研究,以推動藥物早日進入國內的臨床試驗。

cureyoyo網站
秦可佳希望,有關部門能區分用於治病救人的基因治療與違背倫理的基因編輯,不因為個別違規事件,導致罕見病患者失去救治機會。
她也期待,國家能夠盡快頒布孤兒藥鼓勵政策,從醫院到藥審建立罕見病綠色審批通道,寬嚴並濟推動國內基因治療良性有序發展。她呼籲,參考美國NIH對孤兒藥的做法,在罕見病臨床試驗的倫理評審中增加患者家庭(或組織)代表發言,保障那些嚴重危及生命卻無藥可治的患者的生存權。
對秦可佳來說,每天醒來還能看到yoyo的微笑,看到她歡快地跑來跑去,已經很滿足了。
yoyo的父親特地買了一台單反相機,記錄女兒長大的點點滴滴,亦或是,最後的時光。秦可佳卻不敢翻看這些照片,每次她都會忍不住流淚。“我多麽希望時光能夠停留,再給yoyo多一點時間。”

yoyo一家三口在夕陽下 受訪者供圖