專家簡介


李萍,天津市第二人民醫院中西醫結合二科主任,主任醫師,留美博士後,碩士生導師。天津市131創新型人才,中國民族醫藥學會慢病管理分會常務理事,肝病領域雜誌審稿專家。
【病例摘要】
患者××,男, 49歲,因「反覆鞏膜黃染30餘年,加重1月」就診。
30餘年前患者發現鞏膜黃染,伴小便顏色加深,似濃茶水色,無發熱、乏力、食慾下降、噁心、嘔吐、腰痛和周身關節疼痛等癥狀,一直未予重視,偶爾不規則服用中藥,具體成分不詳。黃疸呈間歇性波動,但轉氨酶一直正常。患者平時有雙眼發黃現象,每以感冒、勞累後加重。感冒等好轉後,黃疸有所消退,無發熱、乏力、食慾下降、噁心、嘔吐、腰痛和周身關節疼痛等癥狀,未影響正常工作和生活。近一月來,上腹部不適較頻繁,自服「奧美拉唑、甲硝唑」等藥物,治療效果不明顯而尿色明顯加深,遂來我院就診。既往無肝炎、結核、腎病等病史,無外傷、手術及藥物過敏史。生居本地,不吃生魚,無疫水接觸史,無煙、酒等不良嗜好。
體格檢查:體溫 37℃ 脈搏 82次/分 呼吸 22次/分 血壓 12/8 kPa 發育正常,營養良好,神志清楚,查體合作,自動體位,皮膚鞏膜輕度黃染,全身淺表淋巴結未觸及腫大,未見肝掌、蜘蛛痣,鼻孔通暢,副鼻竇無壓痛,口唇無紫紺,口角無潰瘍,咽扁桃體無腫大,頸軟,甲狀腺無腫大,頸靜脈無怒張,胸廓無畸形,胸骨下段無壓痛,雙肺呼吸音清晰,未聞乾、濕性囉音,心率82次/分,律齊,各瓣膜聽診區未聞及病理性雜音。腹平軟,無腹壁靜脈曲張,全腹無壓痛和反跳痛,肝脾肋下未觸及,肝區無叩痛,移動性濁音陰性,肛門、生殖器無異常,雙下肢無凹陷性水腫,中樞神經系統檢查未見異常。
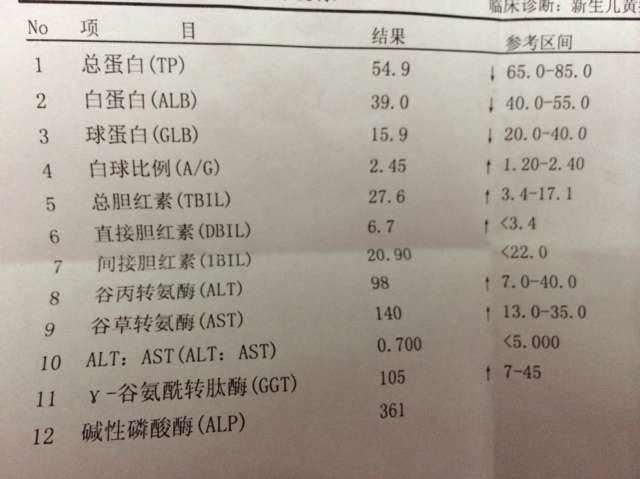
實驗室檢查:血常規 RBC 4.70×1012/L,Hb 150 g/L,PLT 240×109/L,WBC 5.0×109/L,網織紅細胞計數正常,血清肝功能ALT 26 U/L,AST 27 U/L,AKP 50 U/L,GGT 26 U/L,TBIL 60 umol/L,DBIL 20 umol/L,ALB 49 g/L,AFP 2.6 ug/L,膽汁酸正常。血清甲、乙、丙、丁、戊各項病毒標誌物均陰性,HBV-DNA於正常範圍,凝血酶原時間/凝血酶原活動度(PT/PTA)正常,抗核抗體、抗肝腎微粒體抗體、線粒體抗體M2均陰性。肝、膽、脾、胰、腎超聲及CT未發現佔位病變。
【1-住院醫師】
第一次查房住院醫師:
彙報病歷如上。本病歷有如下臨床特點:①中年男性。 ②反覆鞏膜黃染。③肝功能轉氨酶正常,膽紅素升高,以間接膽紅素升高為主。④血常規正常。⑤病毒標誌物檢查陰性。⑥自身抗體陰性。⑦超聲及CT:未見異常。根據上述特點,首先應考慮黃疸查因,因為無明顯的肝細胞炎症,黃疸輕度升高,本例是否應考慮先天性遺傳性疾病,鑒於病人病情複雜,請上級醫師指導。
【1-主治醫師】
第一次查房主治醫師:
黃疸是指皮膚與粘膜因膽紅素沉著而致的黃染。發生黃疸時,血清膽紅素含量常在34.2μmol/L(2 mg)以上。黃疸首先需要與服用大劑量阿的平及胡蘿蔔素等引起的皮膚黃染相區別。後二者的黃染多為單純皮膚髮黃而無鞏膜黃染,血清膽紅素亦不增高。此外,黃疸還應和老年人的球結膜下脂肪積聚相區別,後者黃染在內眥部較為明顯,球結膜多有凹凸不平的斑塊狀分布。
黃疸系一癥狀,常見於以下疾病:(1)傳染病:常見者有病毒性肝炎、壞死後性肝硬變、傷寒病、敗血症(合併細菌性肝膿腫) 以及鉤端螺旋體病、肝結核等。其中以病毒性肝炎、壞死後性肝硬變最為多見。(2)肝膽和胰腺疾患:如肝臟或膽管的腫瘤、膽囊及膽管炎症或結石,以及胰頭癌等。由於壓迫或阻塞膽管,影響膽汁向腸道的排泄而發生黃疸。如果膽管完全阻塞,大便可變成灰白色。(3)中毒性肝炎:肝臟能處理來自胃腸道的毒物、毒素和藥物,將之轉變為無毒的物質排出體外。在處理毒物或毒素的過程中,可以引起肝臟的損害而引起中毒性肝炎。常見引起肝臟損害的毒物、藥物有磷、砷、四氯化碳、氯苯(六六六)以及乙醚、氯仿、巴比妥酸鹽類、氯丙嗪等。(4)嚴重的心臟病和慢性心力衰竭:由於全身血液循環障礙,使肝臟淤血腫大或發生了肝硬變,尤其發生肺栓塞時,易發生黃疸。(5)溶血性黃疸:因某種原因例如錯輸血型不合的血液以及陣發性睡眠性血紅蛋白尿等引起溶血時,由於紅細胞破壞過多而發生黃疸。(6)對膽紅素有先天性代謝缺陷:慢性特發性黃疸、幼年間歇性黃疸。
上述疾病中,臨床上以前兩項最為多見。結合本病史,可以排除以下疾病:(1)病毒性肝炎:起病多有食欲不振、噁心、倦怠等癥狀。肝可觸及,有壓痛。ALT明顯升高。若是乙型肝炎,HbsAg可陽性,鹼性磷酸酶升高,尿色黃。3周後,自覺癥狀好轉,逐漸恢復。(2)藥物性肝炎:有服藥史,可有發熱、噬酸粒細胞增多,停葯後可恢復,再用藥可再發。(3)酒精性肝炎:慢性酒精中毒可發生肝炎,表現為食欲不振、噁心、嘔吐、上腹痛及肝腫大。有時可有發熱,持續2周以上,肝組織有脂肪變性及Mallory透明小體。
綜上所述,本患者先天性黃疸的可能性大。
【1-副主任醫師】
第一次查房副主任醫師:
黃疸的鑒別診斷需與其它疾病一樣,需要有詳細的病史、體檢及其它輔助檢查材料供綜合分析。根據發病機理,黃疸可分為以下四個類型。
(1)溶血性黃疸:分為先天性或後天性 先天性溶血性疾病包括①紅細胞膜缺陷如遺傳性球形紅細胞增多症,遺傳性橢圓形細胞增多症。②戊糖磷酸酶缺乏如丙酮酸激活酶或葡萄糖-6-磷酸脫氫酶缺乏。③球蛋白結構或合成缺陷如鐮形細胞病及地中海貧血。溶血病人在穩定的情況下血膽紅素濃度不超過68.4~85.5μmol/L(4~5 mg/dl),在急性溶血或伴有肝、腎疾病的情況下,血清膽紅素可以很高。後天性溶血性疾病包括①血型不配所致的溶血性貧血,與藥物有關自身免疫抗體及惡性疾病②瀰漫性血管內凝血及溶血性尿毒症③在血透中,由於化學、物理及毒物創傷④陣發性睡眠性血紅蛋白尿 ⑤代謝紊亂。
由於紅細胞在短時間內大量破壞,釋放的膽紅素大大超過肝細胞的處理能力而出現黃疸。血清中膽紅素的增高以間接膽紅素為主。如新生兒黃疸、惡性瘧疾或因輸血不當引起的黃疸,都屬於這一類。可有寒戰、發熱、頭痛、肌肉酸痛、噁心嘔吐等癥狀,尿呈醬油色,有血紅蛋白尿,但尿中無膽紅素。
(2)肝細胞性黃疸:由於肝細胞廣泛損害,處理膽紅素的能力下降,結果造成間接膽紅素在血中堆積;同時由於膽汁排泄受阻,致使血流中直接膽紅素也增加。由於血中間接、直接膽紅素均增加,尿中膽紅素、尿膽原也都增加。肝炎、肝硬化引起的黃疸屬於這類。
(3)阻塞性黃疸:膽汁排泄發生梗阻(可因肝內或肝外病變所致,常見為膽道梗阻),膽中的直接膽紅素反流入血而出現黃疸。在臨床上可檢測到血清中直接膽紅素含量增加,尿中膽紅素陽性而尿膽原卻減少或消失。由於膽紅素等膽類物質在體內瀦留,可引起皮膚瘙癢與心動過緩。膽石症、腫瘤等壓迫膽道導致的黃疸屬於這類。
(4)先天性黃疸:系指由於肝細胞攝取膽紅素障礙,或葡萄糖醛酸基轉移酶(BGT)缺乏使膽紅素結合障礙而引起的一類高膽紅素血症。大多為先天性。根據血清膽紅素的性質將黃疸分為兩類:①以間接膽紅素升高為主②以直接膽紅素升高為主。非結合性高膽紅素血症見於①Gilbert綜合征。②Ⅰ型Crigle-Najjar綜合征。③Ⅱ型Crigle-Najjar綜合征。④Lucey-Driscoll綜合征。⑤旁路高膽紅素血症。結合性高膽紅素血症常見於:①Dubin-Johnson綜合征。②Roter綜合征。③良性家族性肝內膽汁淤積症。
Gilbert綜合征於1901年由Gilbert首先報導,系遺傳性或獲得性葡萄糖醛醯轉移酶活力不足所致。遺傳性病人的家族中約1/2~1/4成員發生黃疸,長期不愈,血清中膽紅素波動在17.1~102.6μmol/L(1~6 mg/dl)之間。Gilbert綜合征又名遺傳性非溶血性膽紅素血症、家族性非溶血性無膽汁尿性黃疸和生理性血內膽紅素增高症等。本病在臨床上主要通過排除法診斷,其主要診斷依據:①慢性間歇性黃疸,有家族史,好發於青少年;②全身情況良好,消化道癥狀不明顯,肝不腫大,膽胰系統均無異常;③實驗室資料可除外溶血性、肝細胞性及阻塞性黃疸;膽紅素代謝試驗有助於診斷。有條件的醫院可行肝穿活檢以協助排除肝臟的器質性病變。本病發病機制尚不十分清楚,可能為:⑴肝細胞葡萄糖醛酸轉移酶活性降低;⑵肝細胞攝取膽紅素能力下降;⑶載體蛋白相對缺乏,使肝細胞攝取和轉運非結合膽紅素過程障礙等。除黃疸外,多無其他癥狀;受涼、過勞、飲酒、感染等可誘發或加重;個別患者在黃疸加深時感乏力、消化不良或腹部不適。本例黃疸加重可能與反覆服用治胃病藥物有關。本病除應注意避免各種誘因外一般不需要特殊治療,如必須用藥時,苯巴比妥口服即可,並且療效可靠。一般認為,苯巴比妥作為酶誘導劑,可能促進葡萄糖醛基轉移酶的合成和增加其活性,增加膽汁流量,有利於黃疸消退。本例半月後膽紅素退至正常出院。臨床上如遇黃疸反覆升高而肝功能其他指標正常患者時應注意與本病鑒別。Gilbert綜合征也稱體質性肝功不良性黃疸,病人的黃疸可在出生時或成年期出現,但最常發生於青年期。罹患者大多為男性,黃疸可持續存在達高齡,但往往隨年齡的增長而逐漸減退。
Crigle-Najjar綜合征Ⅰ型和Ⅱ型Ⅰ型原因是酶缺如,嬰兒生後第二天出現黃疸,嚴重者血清膽紅素可達427.5~769.5μmol/L(25~45 mg/dl).常發生核黃疸,為家族遺傳性疾病,患兒預後不良。Ⅱ型是葡萄糖醛醯轉移酶活力低下,甚至部分缺乏,血清膽紅素<342μmol/L(20 mg/dl)不發生核黃疸,這些病人預後尚可。
Dubin-Johnson又稱為慢性特發性黃疸,為遺傳性結合膽紅素增高Ⅰ型,1954年Dubin等首先報告。Dubin-Johnson綜合征臨床表現特點為長期性或間歇性黃疸。 多數研究表明Dubin-Johnson綜合征血緣相近比率很高,屬常染色體隱性遺傳性疾病,一家可多人發病,病人是Dubin-Johnson綜合征致病基因的純合子,但也有些病人並無家族史。常見於青年人,世界各地均有病例報告。1.可無明顯癥狀;2.輕中度黃疸、尿色加深; 3.右上腹不適或隱痛、乏力、食欲不振、噁心、嘔吐;4.黃疸、肝脾輕度腫大或輕微壓痛。Dubin-Johnson綜合征預後良好,無需特殊治療。 2.苯巴比妥有助於降低血膽紅素。
Dubin-Johnson綜合征,由於毛細膽管對有機陰離子的轉運障礙,致使結合膽紅素從肝細胞向毛細膽管的運轉發生障礙,結果使結合膽紅素反流入血,血結合膽紅素水準增高,病人出現黃疸。Dubin-Johnson綜合征診斷並不困難,但應與Gilbert綜合征、肝炎、梗阻性黃疸,溶血性黃疸等相鑒別,Dubin-Johnson綜合征經過及預後良好,沒有也無需特殊治療,但要盡量避免黃疸加重的誘因如過度疲勞、飲酒、感染、妊娠及口服避孕藥等。
Roter綜合征,1948年Roter首先報送。Rotor綜合征也是血中膽紅素升高以直接型為主。可能由於肝細胞對膽紅素貯藏能力明顯減少所致,肝組織學正常。由於肝細胞的攝取(間接膽紅素)和將結合膽紅素排泌到膽管的作用有先天性障礙,導致間接和直接膽紅素增高。但多不超過200μmol/L,間接膽紅素約佔1/3,直接膽紅素中40%不是膽紅素葡萄糖醛酸酯,而是其他結合膽紅素(如與硫酸結合的膽紅素),血清總膽汁可中度增高,BSP排泄緩慢,但口服膽囊造影劑膽囊顯影良好。
家族性肝內膽汁鬱滯黃疸,亦稱Byler病,1965年Clayton首先報導,在Jacol-Byler的後代中出現了較多的這種病人。為常染色體隱性遺傳病。肝細胞將直接膽紅素和膽汁酸排泌到毛細胞膽管中的功能障礙,導致血清中直接膽紅素增高。肝細胞中有淤膽,毛細膽管中有膽栓,匯管區有淋巴細胞浸潤,偶見肝細胞點狀壞死和纖維化。
在排除了別的引起黃疸的可能性以後才能診斷為Gilbert綜合征。
【2-主治醫師】
第二次查房主治醫師:
肝穿病理顯示:肝小葉結構完整,肝細胞腫脹,胞漿內有微細膽紅素顆粒,少量肝細胞噬酸性變,小葉內偶見噬酸性粒細胞,匯管區大致正常。免疫組化顯示:HbsAg(-),HbcAg (-),Pre-s1(-)。病理診斷考慮為Gilbert綜合征,不排除藥物性肝損傷。
【2-主任醫師】
第二次查房主任醫師:
目前診斷 Gilbert綜合征。該綜合征為常染色體隱性遺傳病。發病率為3%~7%。患者雙親中25%、同胞中50%有血清膽紅素升高。位於染色體2q37膽紅素尿苷葡萄糖醛酸轉移酶基因(UGT1A1)5′端上遊啟動子A(TA)TAA盒結構,其中胸腺嘧啶(TA)的重複序列,正常為6個TA[A(TA)6TAA],本病時插入了2個核苷酸(TA),成為A(TA)7TAA。使UGT1A1基因表達下降,導致膽紅素尿苷葡萄糖醛酸轉移酶活力下調。此外還有(TA)5/6,5/7和7/8等多種變異的等位基因。日本的學者尚報導甘氨酸71精氨酸,脯氨酸229谷氨醯胺,精氨酸367甘氨酸等變異。以(TA)的突變為主要遺傳因素。肝臟對血清內非結合膽紅素攝取和結合能力低下是本病的基本缺陷,其對膽紅素清除值平均僅為正常人的1/3左右。患者肝組織內膽紅素尿苷磷酸葡萄糖醛酸轉移酶活力僅為正常人20%左右,膽汁內膽紅素二葡萄糖醛酸酯比例下降,單葡萄糖醛酸酯的比例上升。除對非結合膽紅素外,部分病人對磺溴酞鈉(BSP)、吲哚氰綠(ICG)和熊去氧膽酸等的攝取也有缺陷。少數病人中雖無溶血表現,但紅細胞壽命縮短。下列方法有助於本病的診斷:①肝活組織標本內葡糖苷酸轉移酶活性降低。②服用苯巴比妥鈉,0.6 g,每日3次,3 天后血清膽紅素濃度可明顯下降或接近正常。③肌餓試驗:進低熱量飲食,即每天1674 J,2天,血清膽紅素可較服低熱卡飲食前上升至少2~3倍。③煙酸試驗:靜脈注射或口服煙酸,可使血清膽紅素濃度增高。本病無需特殊治療,預後良好。苯巴比妥僅能作為診斷試驗用,不能用於治療。具有基因突變的純合子,可以增加新生兒期黃疸的深度及時間,對合併慢性乙型肝炎,慢性丙型肝炎的患者使黃疸加深,急性肝炎恢復期發生輕度黃疸,肝移植時供肝如為純合子則出現黃疸。有報導利福賓士療重度非結合性高膽紅素血症,可以使膽紅素下降。
【診療結局】
本例患者臨床表現、實驗室檢查、肝穿結果都符合Gilbert綜合征。給予保肝及苯巴比妥30 mg,3次/d,口服。治療3 d,黃染癥狀明顯減輕,膽紅素降在正常範圍。隨訪1年,肝功能和肝臟超聲正常。
【經驗總結】
診斷方面 1、臨床表現為慢性間歇性黃疸,全身狀況良好,多因受涼、勞累、感冒、發熱等誘發或加重;2、可無消化道癥狀,一般無肝脾腫大及明顯肝區壓痛,偶伴乏力;3、非結合膽紅素升高為主,肝功正常,尿膽原不增加;4、實驗室檢查排除溶血性黃疸、肝細胞性黃疸及梗阻性黃疸;5、苯巴比妥治療有效,預後良好;6、肝穿刺活檢為診斷的金標準,有關報導飢餓試驗陽性是本病的特異性。本病應注意與溶血性黃疸、肝細胞性黃疸、梗阻性黃疸、脂肪肝、病毒性肝炎、肝硬化相鑒別,還應注意與其他高膽紅素血症CriglerNajjar綜合征、DubinJohnson綜合征和Rotor綜合征相鑒別。
本病預後良好,應積極向患者家屬交代病情變化,消除不必要的顧慮。為避免漏診或誤診此病,應詳細詢問病史,結合患者臨床表現及生化檢查,重視肝穿活檢,綜合診斷,避免給患者帶來精神和經濟負擔。
節選自《疑難及重症肝病查房實錄》一書
TAG: |