神經醫學社區
致力於幫助神經科醫生逐步提高專業水準!

 播放GIF
播放GIF
嬰幼兒癲癇性腦病是一組疾病的總稱,其表現為進行性智力倒退、認知障礙、思維緩慢、言語不清、注意缺陷、行為異常等, 與腦變性病相似。
癲癇性腦病( epifeptic encephalopathy) 是指由癲癇性異常引起的進行性功能障礙,其表現為進行性智力倒退、認知障礙、思維緩慢、言語不清、注意缺陷、學習不能、行為異常等,與腦變性病相似。該病不是一個獨立的疾病,是一組疾病的總稱,其共同特徵主要表現為慢性進行性的神經功能衰退,其主要原因是十分頻繁或嚴重的臨床癲發作和(或)持續大量的癲癇性電活動。
此外,癲癇性腦病具有年齡依賴性,每種疾病都發生在特定的年齡階段,有相對刻板的臨床和腦電圖特徵,並且會隨著年齡的增加發生較大的變化;腦電圖嚴重異常;發作開始前,發育相對正常,發作開始後,神經功能的發育出現延遲、停滯甚至明顯衰退;在腦電圖異常的基礎上可伴或不伴臨床發作;對傳統的抗癲癇藥物反應差;腦電圖特徵與疾病的進程密切相關;局限性的皮質病變可能是全身性癲癇綜合征的原因。
癲癇性腦病的病因及分類
癲癇性腦病多為癥狀性或隱源性癲癇,病因多種多樣,包括:缺氧缺血性腦病;中樞神經系統感染如各種腦膜炎、腦炎、感染中毒性腦病等;代謝性疾病如非酮性高甘氨酸血症、甲基丙二酸血症、丙酸血症、維生素B6缺乏症及門克斯病;遺傳性疾病如結節性硬化、神經纖維瘤病等;皮質發育障礙如半側巨腦、局部皮質發育不良、雙側側室周圍結節性異位、無腦回畸形、巨腦回、腦裂畸形、多小腦回;其他: Sturge Weber 綜合征、腫瘤、孔洞腦、錯構瘤等;病因不明:有部分患者無病因可尋。
新生兒期癲癇性腦病
新生兒期癲癇性腦病包括早期嬰兒癲性腦病伴暴發抑製( EIEE) 又稱為大田原綜合征。早期肌陣攣腦病(early mhyoclonic encephalopathy, EME)。二者均於生後3個月內起病,腦電圖以暴發-抑製表現為主要特徵,病例均出現嚴重的智力障礙和神經系統異常,治療困難,預後不良。
嬰兒早期癲癇性腦病
多數患兒出生早期正常,平均起病年齡3個月(13d--7個月)之後出現難以控制的遊走性局限性發作。可表現為為形式的運動性發作, 如雙眼偏斜凝視、眼瞼、抽動、肢體抖動等,也可繼發全面性發作。發作頻率逐漸增多,最終發展為持續性發作伴精神運動發育進行性倒退,無家族病史。
EEG表現為發作在一側半球內或雙側半球之間移行,累及多個部位。臨床發作與EEG放電在時間與部位上密切相關,發作間期可表現為多種EEG異常。起病後精神運動發育呈進行性衰退,患者肌張力低下,精神萎靡。神經放射及生化檢查無異常發現,無家族性發病。頻繁的發作可能是此病精神運動衰退的唯一原因,預後差。
Ohtahara綜合征
Ohtahara綜合徵發病年齡早,在新生兒期及嬰兒早期,一般在3個月內,多數在1個月內。而且發作形式多為強直性痙攣,散發或成串發作,出現在覺醒及睡眠中。EEG表現為爆發(3-4秒)-抑製(5-10秒),持續出現在覺醒及睡眠中。Ohtahara綜合征多數有腦結構性異常,主要是出生前的腦發育不良。然而,預後不良,均有嚴重NS發育落後,死亡率高,存活病例的臨床和EEG特徵在數月後演變為嬰兒痙攣。治療非常困難。

嬰兒痙攣症
嬰兒痙攣症(IS)起病年齡在2個月-1歲,高峰在4-6個月。80%為癥狀性,可因各種先天性或獲得性腦損傷引起,典型病例表現為特有的三聯征:痙攣發作、腦電圖、高度失律及精神運動發育落後。而且發作常常難以控制,23%-60%以後轉變為Lennox-Gastaut綜合征(LGS)。
嬰兒嚴重肌陣攣癲癇
嬰兒嚴重肌陣攣癲癇(SMEI),臨床特點表現為起病年齡在1歲內,最初表現為發熱誘發的長時間的全身性或一側發生癲持續狀態。患兒發病後有進行性精神運動發有倒退和錐體束征等異常神經體征,特別是語言發育遲緩。無病因可尋,部分患兒有癲癇或熱性驚厥家族史。
在發作間期腦電圖正常,額、中央、頂區可有4-5Hz陣發性節律,一側性發作後顯示背景活動不對稱。1歲以後EEG出現發作間期多導棘慢波發放。全身性的肌陣攣發作時可記錄到多導棘慢波或多棘慢波爆發。散發性肌陣攣、陣攣或局限性發作時EEG可無持續的棘慢波,僅為節律性慢波或與肌陣李無關的散發棘波、棘慢波。癲癇發作對各種抗癲治療反應均不好,長期預後較差。
兒童期癲癇性腦病(LGS)
LGS起病多在3-5歲,病因包括多種先天性或獲得性腦病變,少數為隱源性病因,男性多於女性。臨床特點表現為:①頻繁的、形式多樣的癲癇發作,包括強直發作、不典型失神發作、失張力發作、肌陣攣發作等。其中,強直發作是LGS最具有特徵性的發作形式見於92%的患兒且很少見於其他癲綜合征;②腦電圖有彌散性1.5- 2.5Hz棘慢波,97%的患兒可見廣泛性棘波節律或快波節律爆發,是LCS最具特徵的腦電圖改變;③智力發育落後,病程常為進行性。LCS治療困難,是常見的難治性癲癇之一。
Landau-K leffner綜合征( LKS)又稱為獲得性癲癇性失語,主要臨床特點為兒童期獲得性失語,失語出現的年齡為3 -12歲,患兒起病前發育正常,多呈亞急性起病,典型者表現為言語聽覺失認,癥狀呈波動性變化,伴精神行為異常;腦電圖以顳區為主的樣放電,睡眠期可出現廣泛或局限性的頻發棘慢複合波,常呈持續性出現,為L.KS實驗室檢查唯一的異常。癲癇發作可見於70%的患兒,包括全面和(或)局限性發作,其臨床發作和EEG異常常呈良性經過,預後大多良好,但常遺留神經心理學損傷。
癲癇伴慢波睡眠期持續棘慢波(CSWS)
CSWS是一種少見的年齡相關性的兒童癲癇,最突出的特徵是睡眠中的電持續狀態和高級皮層功能損傷。起病年齡6-14歲,高峰年齡8歲,男童多見。腦電圖以慢波睡眠期持續性棘慢波發放為特徵,以額區或前頭部為主。80%有局限性和(或)全面性癲癇發作,但無強直發作。癲癇發作一般呈良性演變過程,但神經心理學方面的預後不好,患兒有廣泛的認知障礙、智力倒退和行為問題。
Rasmussen綜合征(RS)
Rasmussen綜合征是一種起源於新皮層的部分性癲癇,病因尚未完全明確,目前多認為是一種自身免疫性疾病,病理特點是一側大腦半球限局性慢性炎症。臨床有3個特點:①逐漸加重的局限性運動性癲發作,常發展為持續性局限性癲癇(epilepsia partialis continua, EPC), 抗癲藥物不能滿意控制發作;②逐漸加重的進行性偏癱;③漸進性的神經精神心理學損害。
腦電圖發作間期可為一側半球局灶性、一側半球多灶性或雙側半球多灶性癲樣放電,均可伴雙側同步放電,少數僅表現為雙側同步放電;發作期EEG放電與局部肌陣攣性抽動沒有很好的相關性。影像學檢查早期正常,以後可見一側半球萎縮性改變。50%患兒有腦脊液異常,包括細胞數和蛋白輕度增高,也可出現寡克隆或單克隆帶。
癲癇性腦病的診斷
癲癇性腦病的診斷應依據全面而詳盡的臨床資料和有關的實驗室檢查,包括臨床癥狀與體征,每種癲癇性腦病都有相對特異的臨床表現,可通過患者的癥狀、體征以及它們的共同特徵予以初步診斷。此外包括腦電圖檢查和神經影像學檢查。其中,神經影像學檢查包括CT、MRI檢查可發現各種先天性的大腦發育不良、畸形、炎症、腫瘤等,為診斷和治療提供幫助。
功能影像學檢查,比如PET、SPECT等,很多病例通過常規的臨床腦電圖及MRI等檢查未發現異常,但通過功能影像學檢查卻可能有陽性發現。神經心理學檢查是癲癇性腦病的患兒大多有精神運動發育障礙,智力及認知功能損害,因此神經心理學檢查是不可缺少的,可為診斷、治療及預後提供依據。
癲癇性腦病的治療原則為病因治療,控制癲癇發作,使異常的腦電圖恢復正常,神經心理學康復治療。癲癇性腦病常系難治性癲癇,其治療應根據個體特點採用多方面措施,依靠多學科協作來進行,是一個包括藥物、激素,飲食、心理、外科等方面的綜合治療過程。針對病因進行特異性治療是癲癇治癒的關鍵,而癲癇性腦病多系癥狀性癲癇,因此應盡量尋找病因,給予相應的治療。
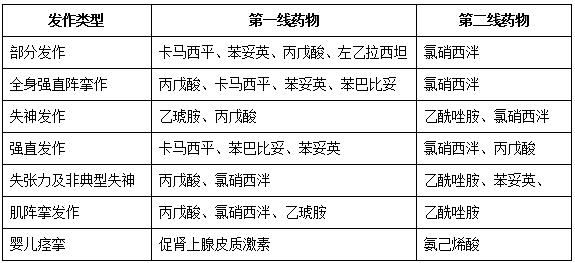
抗癲癇藥物(AEDs)治療
抗癲癇藥物治療包括傳統AEDS和新型AEDS。目前國內應用較多的聯合用藥方式有丙戊酸鈉+拉莫三嗪、丙戊酸鈉+托吡酯、丙戊酸鈉+卡馬西平、拉莫三嗪+托吡酯等。一般主張兩種抗癲癇藥物聯用,最多不要超過三種。新型抗癲癇葯是治療難治性癲癇的主要選擇藥物,如拉莫三嗪、托吡酯、左乙拉西坦、奧卡西平、加巴噴丁、氨已烯酸、苯丙氨酯、唑尼沙胺等。
類固醇/ACTH治療
皮質激素治疔癲癇的作用機制尚不十分清楚,推測可能與以下機制有關。1、抗感染、免疫調節與免疫抑製作用;2、反饋抑腎上腺皮質激素釋放激素 ( CRH)對動物腦室內注入CRH,可致嬰兒痙攣症樣發作,ACTH通過負反饋抑製CRH分泌,起到止驚作用;3、ACTH促進脫氧皮質酮分泌,脫氧皮質酮的代謝產物3α,5α-別四氫脫氧皮質酮作用於γ-氨基丁酸A受體(GABAA)受體,使氟離子通道開放,產生抗驚厥作用;4、促進腦成熟:ACTH可增加腦脊液神經生長因子水準,促進腦成熟;5、直接作用於中樞神經系統受體:體外實驗顯示其可降低海馬錐細胞的興奮性;增加腦餚液GABA和5-羥色胺,降低N-乙醯天門冬氨酸水準;6、皮質激素具有的糾正低血糖、降低細胞內糖、改變細胞內外的離子梯度、減少腦組織內含水量、及酶修復等功能,對控制驚厥可能起一定作用。
靜脈注射兔疫球蛋白
許多耐藥性的癲癇性腦病如West綜合征、LGS、LKS、 嚴重肌陣攣性癲癇,Rasmussen綜合征(RS)等對靜脈注射疫球蛋白有效,但易複發。不過由於副作用小,對AEDs無效的患者可考慮使用。
生酮飲食控制發作的作用機制
生酮飲食(ketogenic diet)是一種高比例脂肪、適量蛋白質和低碳水化合物的飲食,脂肪:(蛋白質+碳水化合物) =4:1,它將身體的主要代謝能源從利用葡萄糖轉變為脂肪,通過肝臟代謝產生酮體,從而導致機體的一系列反應。 這種用含脂肪比例高、蛋白質和碳水化合物比例低的飲食配方,通過脂肪分解代謝產生酮體,模擬身體對飢餓的反應治療癲癇等疾病的方法稱為生酮飲食療法。
在進食過程中,葡萄糖通過促進葡萄糖轉運載體進入腦部,在進食過程中,脂肪酸為肌肉和其他組織提供能量,但它不能進入腦部,由脂肪酸產生的酮體和肝臟中的生酮氨基酸通過轉運載體,進入大腦為其提供另一種能量,可以產生對腦部的抗驚厥作用,其具體抗驚厥機制還不清楚。一般認為有以下幾方面:1.改變腦的能量代謝方式;2.改變細胞特性,降低興奮性和緩衝癲癇樣放電;3.改變神經遞質,突觸傳遞,神經調質的功能;4.改變腦的細胞外環境,降低興奮和同步性。
生酮飲食的缺點,不適於遺傳代謝性疾病;副作用包括胃腸道癥狀、睏倦或嗜睡、低血糖。睏倦或者嗜睡、過度酮症;遠期副作用包括:腎結石、低蛋白血症、高脂血症、便秘、生長障礙、骨代謝障礙;成本高、製備麻煩。
此外,還有外科治療。手術治療一方面主要針對結構性異常,如對那些局部皮質發育不良、大腦發育畸形、腫瘤、腦積水等,切除病灶後可使部分患者的癲癇得到控制。另一方面對通過PET、SPECT等功能影學檢查發現的致癇灶採用手術切除有時亦會收到意想不到的效果。
綜上所述,癲癇性腦病是一組疾病的總稱,其共同特徵主要表現為慢性進行性的神經功能衰退,其主要原因十分頻繁或嚴重的臨床癲癇發作和(或)持續大量的癲癇性電活動。具有年齡依賴性,每種疾病都發生在特定的年齡階段,有相對刻板的臨床和腦電圖特徵,並且會隨著年齡的增加發生較大的變化。發作開始後,神經功能的發育出現延遲、停滯甚至明顯衰退。對傳統的抗癲癇藥物反應差,預後不良。