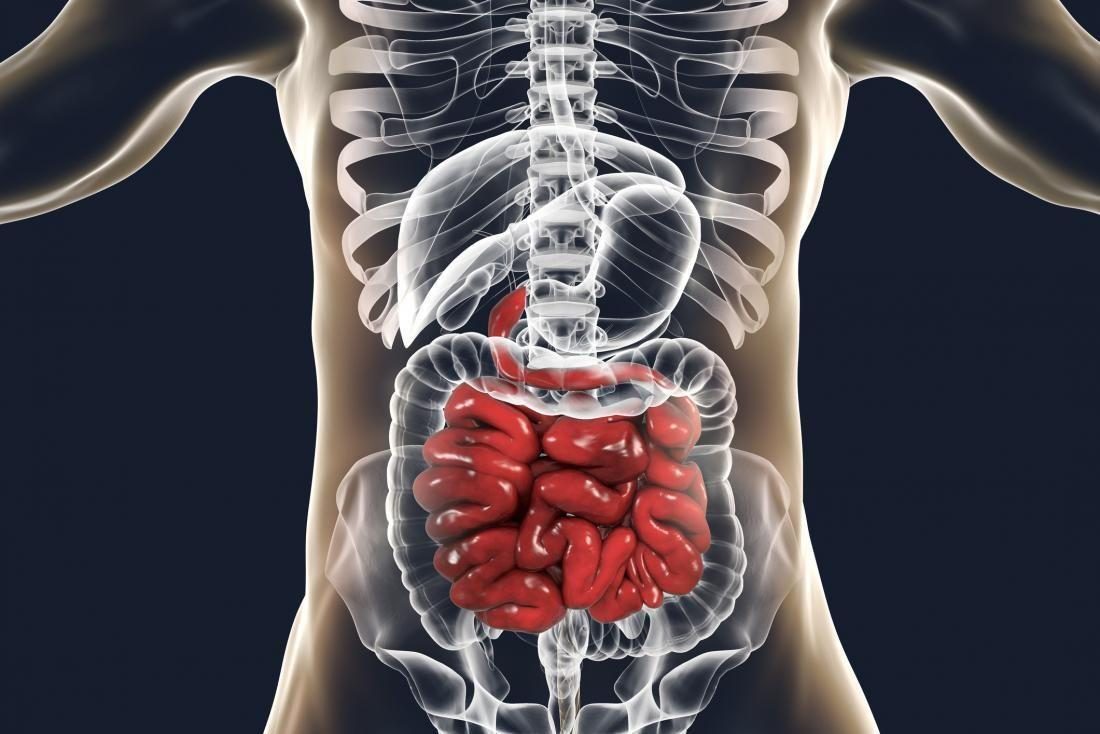
細胞中腫瘤抑製因子的丟失,基質細胞驅動胃腸道息肉的生長
非激活導致腫瘤抑製基因stk 11(絲氨酸-蘇氨酸激酶11)功能喪失的體細胞突變,編碼肝激酶B1(LiverkinaseB1)的蛋白在sev中經常發生。 散發性癌症,尤其是肺癌、胰腺癌和女性生殖腫瘤。此外,STK 11中的遺傳性雜合子生殖系突變導致了PJS,一種癌症前發性綜合征。 宗教錯位綜合征。PJS的一個特點是許多良性胃腸道(GI)錯構瘤息肉的生長,以及多個器官發生惡性腫瘤的風險升高。研究 小鼠的PJS模型指出LKB 1缺乏的基質,而不是上皮,是導致牙齦息肉形成的因素。基質由成纖維細胞、平滑肌細胞組成。 支持上皮組織的細胞外基質和基底膜。
然而,LKB 1缺乏的基質細胞如何觸發息肉的形成尚不清楚。最近的兩項研究揭示了PJS息肉形成的機制。發現T細胞STK 11單個等位基因的缺失足以促進胃腸道息肉的形成。同樣,Ollila等人的一項研究。證明了雜合子l STK 11在胃基質細胞中的OSS足以促進胃腸道息肉的形成。在這兩項研究中,轉錄譜顯示炎症細胞因子的上調參與了促進。間質和正常胃腸上皮的擴張和過度生長。通過發現由於T細胞或基質細胞丟失而導致間質室炎症導致息肉 這些研究突出了炎症信號如何深刻地改變微環境和促進腫瘤的形成。

PJS患者的GI息肉含有多種細胞類型,它們都遺傳了STK 11的雜合子突變,並以平滑肌核心為特徵,一個擴大的間質室和增生上皮。Poffenberger等人STK 11等位基因在不同細胞間隔中被條件缺失的小鼠模型 要了解哪些細胞有助於息肉的形成。他們觀察到Gi上皮細胞STK 11雜合子缺失並不能促進息肉的形成。出乎意料的是,他們發現 STK 11在造血細胞中的雜合缺失足以驅動與STK 11雜合子小鼠和w雜合子小鼠組織學上難以區分的GI息肉的生長。 HICH也類似於PJS患者的息肉。此外,他們發現在B細胞中STK 11雜合子的丟失與息肉的形成無關,而T細胞中STK 11的雜合子丟失則不起作用。 LS可促進腸息肉病的發生。補充這些發現,Ollila等人。結果表明,STK 11在胃基質細胞中的雜合缺失足以誘導pjs樣多聚體的形成。

LKB 1的丟失如何導致息肉的形成?LKB 1的活性對細胞的極性、生長和代謝產生多種作用,包括對細胞能量脅迫的反應。 磷酸化,從而控制腺苷一磷酸(AMP)依賴性蛋白激酶(AMPK)的激活。AMPK對雷帕黴素哺乳動物靶標活性的負調節作用 複合物1(MTORC 1),細胞生長和增殖的中央調節因子。因此,無論是lkb 1的表達還是ampk的激活,都會導致mtorc 1信號的上調。 在許多情況下,包括PJS息肉病和散發性LKB 1缺失的腫瘤中,被認為有助於腫瘤的生長。先前的研究發現上皮複合物中mTORC 1信號增強。 PJS息肉的克隆,以及Poffenberger等人的研究。當LKB 1僅在T細胞中被刪除時,也觀察到了這一事件。令人驚訝的是,T細胞特異性消融ampk的小鼠沒有發育。 鼻息肉此外,T細胞中STK 11和mTOR的同時缺失仍可導致GI息肉的發生。

這些結果表明AMPK在這些模型中,T細胞中的mTORC 1對PJS息肉的發育是可有可無的,而LKB 1依賴的信號通路必須負責息肉的生長。同樣,Ollila等人。 據報導,Gi間質室AMPK的失活並沒有引起息肉病。來自LKB 1缺乏的T細胞或胃基質細胞的什麼信號足以驅動息肉的生長?Poffenberger等人發現T細胞STK 11雜合子丟失誘導活化 F CD4和CD8 T細胞在腸系膜淋巴結中,這些T細胞、巨噬細胞和中性粒細胞明顯浸潤到息肉的微環境中。雜合表達 小鼠STK 11,特別是T細胞或基質細胞,導致大量炎性細胞因子的分泌,其中包括白細胞介素-6(IL-6)和白細胞介素-11(IL-11)。 Sly與胃腫瘤的發生有關,細胞因子的增加伴隨著jak-stat(Janus kinase-信號轉導和轉錄激活劑)si的過度激活。在基質室叮咬,這會導致炎症和癌症。

此外,用JAK的抑製劑AZD 1480處理可使STK 11雜合子小鼠的STAT 3激活失活,並顯著抑製息肉的生長。同樣,Ollila等人。發現 在激活的JAK-STAT 3信號通路上,基質細胞中的信號驅動息肉的形成,而JAK抑製劑ruxolitinib(已被臨床批準用於治療骨髓增生疾病)具有治療作用。 TiAl在PJS小鼠模型中的作用。綜合起來,出現了一種模型,即T細胞或胃間質細胞缺乏LKB 1會產生促炎微環境,隨後產生炎症微環境。

如果不通過降低AMPK功能,LKB 1缺乏如何導致炎性細胞因子的產生和促進?除了激活AMPK外,LKB 1還能磷酸化並激活 另外12種AMPK相關激酶家族,Poffenberger等人未探討其在T細胞解除管制中的潛在作用。特別是Ollila等人。報告Ampk家族成員SIK 1(鹽誘導激酶1)、Mark1(MAP/微管親和力調節激酶1)或Mark4的表達降低,可誘導成纖維細胞產生IL-11。 有趣的是,這些AMPK家族中的一個或多個成員可能介導在人和小鼠PJS息肉中觀察到的促炎細胞因子風暴的產生。

Poffenberger等人的發現。和Ollila等人。構成一個重要的例子,說明遺傳性家族腫瘤抑製基因的單倍體功能如何導致炎症。 癌症綜合症。與LKB 1缺乏PJS相似,其他抑癌因子雜合性缺失也能促進良性腫瘤生長,包括上皮細胞、基質細胞和免疫細胞。 細胞成分,混合系腫瘤雜合子丟失的例子包括家族性腺瘤性息肉病中的APC(腺瘤性息肉病)丟失,家族性幼年性息肉病(Fj)中缺失Smad 4型。PTEN(磷)TSC 1(結節性硬化複合體1)和TSC 2(結節性硬化症)、VHL(vonHippel-Lindau抑癌基因)、vonHippel-Lindau綜合征(vonHippel-Lindau)和NF1(Neur) 神經纖維瘤病1型。如Poffenberger等人所證明的,免疫細胞成分參與了良性腫瘤的形成。小鼠模型中的回聲觀測 如fjp,T細胞中smad 4的丟失可調節炎性細胞因子,導致胃腸道息肉的生長,並導致1型神經纖維瘤病(nf1-異種)。 合肥肥大細胞(一種炎症免疫細胞)向神經微環境分泌促炎細胞因子,這是神經纖維瘤生長所必需的。

會很幼稚的 靜坐觀察除pjs,fjp和神經纖維瘤外,其他家族性癌症綜合征中,免疫系統的參與是否也會促進良性腫瘤的生長。 S型1或散發性實體瘤伴LKB 1缺失。Poffenberger等人的發現。還提示JAK抑製劑可能是治療PJS患者胃腸道及其他腫瘤的新方法。 治療方案寥寥無幾,對靶向JAKSTAT 3信號在散發性LKB 1腫瘤中的參與和治療潛力的未來探索也是必要的。