文/田雨
編/ HLR
前幾天看到一個虐心的六字小說:「轉賣:嬰鞋,全新」,作為一個媽媽,我第一時間感受到了作者難以宣洩的傷痛。每一個曾經孕育過生命的母親大約都體驗過這樣的焦慮和擔憂:我的孩子到底能不能健康地發育完成。按照常規,我本人也是在第16周的時候接受的唐氏篩查,結果提示21三體高風險,醫生的建議是做染色體核型分析,那是2011年,染色體核型分析需要等一個半月才能出結果,雖然最終核型分析的結果令人欣慰,可是這一個半月的心理感受我一輩子都忘不掉。
醫學始終在進步,要問21世紀,醫學領域最值得期待的技術突破有哪些,很多人可能首先會想到基因治療,對生命本源孜孜不倦的探索,使得我們日益了解自身,同時也希望能從根本上治癒那些傳統療法束手無策的疾病,比如基因變異導致的許多罕見病。據統計,約有50%的罕見病在新生兒期或兒童期即可發病,病情進展迅速,死亡率高,給患者家庭帶來極大痛苦。預計我國罕見病的病人數將超過1000萬。基於罕見病藥物研發的高風險性,許多國家或地區都對罕見病的研發給予了政策上的支持。今年6月8日,國家衛生健康委員會等五部委聯合制定的《第一批罕見病目錄》正式發布,共有121種疾病被收錄其中。這被業內視為罕見病保障歷史性的突破,有望推動罕見病的基礎研究和臨床研究,解決無葯可治或有葯難尋的難題。
說到有葯難尋,巧合的是,就在罕見病目錄發布後的一個月,電影《我不是葯神》熱映,輿論嘩然,和影片中講述的白血病患者一樣,罕見病這個群體也在經歷同樣的苦難,許多治療罕見病的孤兒葯也和「格列衛」一樣受市場小、研發風險高的因素製約而價格昂貴或不能迭代更新。新政策的頒布在很大程度上能加速海外孤兒葯在國內上市並且進入醫保覆蓋範圍。
這篇文章就121種罕見病之一的遺傳性低磷酸酯酶症及目前的各種治療藥物研發情況進行一下詳細介紹 。
1
概要
低磷酸酯酶症(Hypophosphatasia,HPP)是一種罕見的代謝性骨病,發病率約1/10萬。1948年,由John Campbell Rathbun提出該病的命名。這是一種罕見的遺傳性疾病,是由 ALPL 基因突變引起其編碼的組織非特異性鹼性磷酸酶(tissue nonspecific alkaline phosphatase,TNSALP)活性降低所致,其特徵在於骨骼和牙齒的鈣化缺陷,這些缺陷源於骨骼和牙齒無法正常吸收鈣和磷等礦物質進而發生骨骼變軟和骨骼畸形,並易發生骨折;而牙齒的礦化缺陷則可導致牙齒過早脫落。HPP的癥狀因人而異,同一家庭的成員之間也是如此。HPP有六種主要的臨床形式,極其嚴重的可引起胎兒在子宮內就死亡,相對較輕的會在嬰兒期過早喪失乳牙但不出現骨骼異常。
鹼性磷酸酶(alkaline phosphatase ALP)是一種通過磷脂醯肌醇聚糖的羧基末端錨定到細胞膜上的磷酸單酯酶。人類 的ALP有四種同工酶:腸型、胎盤型、胚細胞型和組織非特異型(TNSALP)。TNSALP 廣泛存在於肝臟、骨骼和腎臟組織等,其主要生物學功能是水解細胞外底物無機焦磷酸鹽(PPi)、磷酸吡哆醛(PLP)和磷酸乙醇胺(PEA)。產生無機磷酸鹽,促進成骨細胞的礦化作用。如果 TNSALP 功能缺陷,則可導致上述底物在體內積累,間接導致體內鈣水準升高和骨骼鈣化不足,骨骼、牙齒等硬組織礦化遲緩。通常,TNSALP酶活性與HPP的嚴重程度相關,體內殘餘酶活性越低,臨床癥狀和體征越嚴重。
2
HPP的癥狀和體征
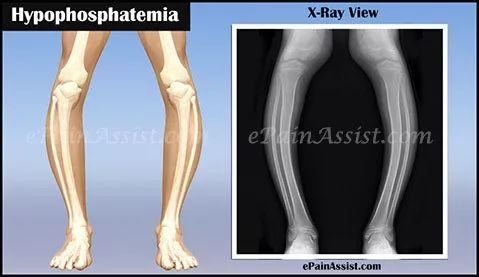

HPP是一種極易變異的疾病。基於臨床癥狀和發病年齡已經確定了六種主要的臨床形式。分別是圍產期致死型,圍產期良性型,嬰兒型,兒童型,成人型和牙齒型。通常,這些不同形式的HPP的嚴重性與體內殘留的鹼性磷酸酶活性相關,酶活性越低所導致的臨床表現越嚴重。此外要注意受影響的個體可能沒有下面討論的所有癥狀,也就是說每個病例實際上都是獨特的。有些孩子會在嬰幼兒期出現嚴重的併發症;?而有些患者則只有輕微的癥狀,並且可能會在成年後有所改善。
圍產期致死型HPP與鹼性磷酸酶活性的顯著降低有關,胎兒骨骼存在嚴重的礦化損害,在子宮內無法形成正常的骨骼形態。不同病例骨骼畸形可能會有所不同,但最常出現的是短而弓形的手臂和腿以及發育不全的肋骨。有些情況下妊娠過程以死胎結束;有些情況下,受影響的胎兒可出生並存活數日,但由於胸部畸形和肺發育不全而最終呼吸衰竭。
圍產期良性型HPP患兒出生前通過超聲檢查可以明顯看出骨骼畸形,但骨骼畸形在出生後會有所改善,最終可能是類似於出現從嬰兒型HPP到牙型HPP個體的癥狀和體征。
嬰兒型HPP在出生時可能沒有明顯的異常,但癥狀可以在出生後前六個月內的任何時間變得明顯。最初的癥狀只是與年齡和性別不相符的體重增長緩慢,被稱為「發育停滯(failure to thrive)」。一些受影響的嬰兒後期表現出顱骨早期融合(顱縫早閉),這可能導致頭部出現不成比例的長寬比,並與腦脊液壓力增加有關,這種情況被稱為「顱內高壓」。顱內高壓可引起頭痛,視盤腫脹(視乳頭水腫)和眼球膨脹(眼球突出)。患病嬰兒還會在發育過程中伴隨著骨骼軟化和生長板異常引起腿部特徵性彎曲變形以及手腕和踝關節增大等骨骼畸形,可出現骨骼疼痛。有的嬰兒也可能在胸骨和肋骨出現畸形,這使得他們易患肺炎、不同程度的肺功能不全和呼吸困難等呼吸系統併發症,嚴重時出現導致危及生命的呼吸衰竭。部分嬰兒肌張力明顯降低,主要歸因於血液中鈣水準升高(高鈣血症)。高鈣血症同時還可引起嘔吐,便秘,虛弱和餵食不良。而高鈣血症帶來的鈣的代謝壓力增加,最終還會導致腎臟損傷。在極少數情況下,還可能會發生維生素B6依賴性癲癇發作。有些患兒到了兒童早期礦化會有自發的改善,但身材矮小和骨骼畸形則會持續終生。
兒童型HPP變異很大,但總的來說不如嬰兒型嚴重。患病兒童有時可能患有顱縫早閉並出現顱內高壓癥狀,類似佝僂病的骨骼畸形也會在2至3歲時變得非常明顯。可能會出現骨骼和關節疼痛,易發骨折。通常還會有一顆或多顆乳牙過早脫落。有些孩子身體虛弱,行走時間延遲,當他們學會走路時,會有明顯的蹣跚步態。有文獻報導,隨著年齡增長骨骼癥狀會自發緩解,但這種癥狀可能在中年或成年後期再次出現。
成人型HPP的特點是存在更加多樣的癥狀。受影響的個體患有骨軟化症,有些人在童年時期有過佝僂病的病史,或者過早失去了乳牙。患有成人HPP的個體可能會出現骨折,特別是腳部的應力性骨折或大腿的假性骨折。反覆骨折可導致慢性疼痛和衰弱。脊柱骨折似乎不太常見,但也偶有發生。骨痛是比較常見的併發症。由於鈣晶體(鈣化性關節炎)或稱為軟骨鈣質沉著症的病症,一些患病的成人在某些關節附近或周圍發生關節炎症和疼痛,其原因在於關節軟骨內鈣晶體的積聚,會損傷關節。關節疼痛嚴重(被稱為假痛風)。受累的成年人可能會過早失去恆牙。
牙型HPP的特點是兒童時期乳牙早失,或青年期恆牙異常脫落。牙齒問題是一個孤立的存在,不會出現佝僂病常見的生長遲緩和骨骼畸形。但這一型 HPP卻容易被口腔醫生忽略,不去做血清ALP的檢測,常常被誤診為侵襲性牙周炎。
3
病因

HPP由ALPL基因的突變引起,ALPL是唯一與HPP有關的基因。ALPL 基因位於人染色體 1p36.1-p34,在基因組中的跨度約為 50kb,共 12個外顯子,其編碼的人類 TNSALP 由 524 個氨基酸組成。低磷酸酯酶的發生就是由於 ALPL 基因突變引起 TNSALP 活性下降,進一步引起骨骼及牙齒髮育缺陷及礦化異常。
目前文獻報導了275個 ALPL 基因突變類型均可導致低磷酸酯酶,其中,大約有 80% 為錯義點突變,10% 為缺失突變,4% 為剪切突變,3% 為無義突變,這些突變可以以常染色體隱性(只有純合子時才顯示病狀)或常染色體顯性方式遺傳。圍產期和嬰兒型HPP以隱性方式遺傳。兒童型HPP可以是隱性遺傳或顯性遺傳。成人型和牙型通常是顯性遺傳,但在極少數情況下可以作為隱性性狀遺傳。
4
流行病學
HPP以相同的比例影響男性和女性。在全球各民族中均可見 HPP 患者,但患病率在不同地區差異較大。在加拿大,重型 HPP 患病率約為 1:100,000,在與世隔絕的加拿大門諾派教徒中患病率高達 1:2500,HPP在日本也相對普遍,但在黑人個體中比較罕見。由於很多病例可能未被診斷或誤診,因此很難確定普通人群中HPP的真實頻率。
5
類似疾病
以下疾病的體征和癥狀可能與低磷酸酯酶症相似。比較它們的特徵對鑒別診斷有用。
成骨不全症(Osteogenesis imperfecta OI)是一組罕見的影響結締組織的疾病,其特徵為骨質脆弱、藍鞏膜、耳聾、關節鬆弛,是一種由於間充質組織發育不全,膠原形成障礙而造成的先天性遺傳性骨胳病。像HPP一樣,具體癥狀和體征在個體之間差異很大。OI的嚴重程度在同一家庭的個體之間也有很大差異。OI可能是輕微的或嚴重的。按病情輕重,可分為產前型和產後型。產前型最嚴重,出生時即有癒合的或剛發生的骨折,易形成死胎或出生不久死亡。產後型較輕,有的到兒童期才發生第一次骨折。有的未成年患者平均每年骨折一次。
維生素D缺乏性佝僂病是一種在嬰兒期或童年 時期常常表現得很明顯的病症,其原因是營養不良引起的維生素D不足,缺乏暴露於陽光下或腸道不能充分吸收營養素(包括維生素D)所致。人體要從食物中吸收鈣和磷離不開維生素D,因此,足夠的維生素D對於適當的骨骼發育和生長至關重要。維生素D缺乏性佝僂病的主要癥狀包括骨質軟化,生長緩慢和虛弱。這種疾病在有些國家比HPP更常見,尤其在世界某些陽光不足的地區非常普遍。
X連鎖低磷酸鹽血症(X-linked hypophosphatemia,XLH)是一種罕見的遺傳性疾病,其特徵在於腎臟中磷酸鹽轉運受損和維生素D活性改變。由這些損傷導致的低血磷水準(低磷酸鹽血症)可導致佝僂病或骨軟化症。在兒童中,生長速度比正常情況慢,經常導致身材矮小,同時由於骨骼逐漸軟化而導致的腿部彎曲變形也是主要體征。在成人中,則由於生長板不再開放,以致骨軟化症成為主要問題。X連鎖性低磷酸鹽血症是由PHEX突變引起的,基因位於X染色體上。它是低磷血症最常見的可遺傳形式(發病率約為1 / 25,000新生兒)。然而,也存在罕見的常染色體顯性遺傳和隱性形式的低磷血症。
6
HPP的診斷


上述各種各樣的疾病可能有一些癥狀與HPP患者相似,那麼如何明確診斷?
HPP的診斷基於特徵性癥狀和體征的識別,詳細的患者病史,全面的臨床評估以及各種實驗室檢查,包括常規X射線和生化檢測以及ALPL基因的突變分析。
患者首診往往是因為出現了相關的骨骼癥狀或體征,在最嚴重的HPP病例中,特別是圍產期和嬰兒期,X線檢查可以揭示骨內的病理變化。然而,除了熟悉該疾病的放射科醫師之外,這些改變可能會被誤診。不過除了極少數具有正常活性水準的假性低磷酸酯酶血症的個體,患有HPP的個體往往都有血清鹼性磷酸酶活性的降低,包括一些HPP遺傳攜帶者,但不發展任何癥狀的個體也可能具有低血ALP水準。
因此,生化檢查在HPP診斷中起著很重要的作用,需要注意的是,血清ALP活性的範圍隨年齡而變化。健康兒童的ALP水準通常比健康成人高。如果進行測試的實驗室在其報告中僅給出成人ALP活性的正常範圍,則可能會錯過兒童HPP的診斷,因為兒童的ALP活性將被錯誤地認為是正常的。通過測量血清維生素B6水準也可以進一步支持HPP的疑似診斷。因為PLP(維生素B6的活性形式)通常由TNSALP分解,患有HPP的個體血液中的吡哆醛5"-磷酸(PLP)水準會有明顯升高,即使是HPP輕微的個體或者沒有出現任何癥狀的HPP遺傳攜帶者,PLP都會升高。
分子遺傳學檢測可以有力的支持HPP的診斷。分子遺傳學檢測可以檢測已知會導致疾病的ALPL基因的突變,尤其適用於臨床表現以及實驗室檢測對既往生育 HPP 嬰兒的孕婦無法確診時,突變位點的篩查是比血清 ALP活性檢測更可靠的指標,隨著成本的降低,分子遺傳學檢測有望成為專業實驗室的常規診斷服務。但也應該注意文獻中沒有報導過的新的突變類型的發生。
7
HPP的治療
1)一般療法
2015年,美國食品和藥物管理局(FDA)批準Strensiq(asfotase alfa),這是第一個被批準用於圍產期,嬰兒和青少年HPP治療的藥物治療[1]。所有兒童期發病的HPP患者(無論接受治療時年齡多大)都可以接受皮下注射這種骨靶向形式的TNSALP替代治療。該葯是由位於美國的Alexion Pharmaceuticals 公司開發的,該公司一直專註於罕見疾病療法的開發研究。產品包括治療陣發性睡眠性血紅蛋白尿(PNH)、非典型溶血尿毒綜合征(aHUS)和抗乙醯膽鹼受體抗體陽性的全身型重症肌無力藥物Soliris(eculizumab)以及溶酶體酸脂酶缺乏治療藥物Kanuma (sebelipase alfa)。




2)支持治療
針對可能因個體而異的特定癥狀和併發症,治療可能需要專家團隊的協調努力,兒科醫生,矯形外科醫生,牙科專家(如兒科牙醫),疼痛管理專家和其他醫療保健專業人員需要系統全面地規劃治療。
可以給予非甾體抗炎葯(non-steroidal anti-inflammatory drugs,NSAID)來治療骨和關節疼痛。NSAIDs在使用時需要小心和監測,因為它們會引起副作用(例如它們會傷害胃和腎),特別是當給予過量劑量和長時間使用時。如果顱縫早閉引起顱內壓,可能需要手術來緩解壓力。
維生素B6可以幫助控制嚴重受影響嬰兒的特定癲癇發作。血液中鈣(高鈣血症)水準升高的嬰兒可以通過飲食鈣限制,水合作用,某些利尿劑和降鈣素來治療,但高鈣血症通常難以控制,通常它發生在病情較嚴重的患者中。
建議從早期開始定期進行牙科護理。在某些情況下,可能會建議進行物理治療和作業治療。此外,這類患者在接受牙科種植治療時也要特殊注意,其成功率會受到影響。
重複長骨骨折的成年人可以通過骨科內固定來治療,骨科醫生通過骨的中心開口放置一根金屬棒,使其更穩定和更強壯。專門為足部設計的特殊醫療裝置(足部矯形器)可供成年人用來幫助治療足部骨折。
患病個體應該避免使用雙膦酸鹽(一類用於治療其他骨骼疾病如骨質疏鬆症的藥物)。這些藥物可能會使HPP惡化或在未確診HPP的個體中引起癥狀加重。常見的雙膦酸鹽藥物包括阿侖膦酸鹽,伊班膦酸鹽,帕米膦酸鹽,利塞膦酸鹽和唑來膦酸鹽。
遺傳顧問對患者及其家庭都是有益的,尤其是計劃生育更多子女的家庭,通過實驗室檢測明確突變位點和產前診斷都是十分有必要的,對患有HPP的兒童和整個家庭的心理社會支持也可能有所幫助。
3)研究中的療法
國外文獻報導了兩例沒有親屬關係的女嬰[2],她們均患有危及生命的HPP,在接受了骨髓移植-造血乾細胞移植之後癥狀得到了改善了,其中一個也接受了骨碎片和培養的成骨細胞治療,根據醫學文獻,雖然沒有對這些程式進行更正式的研究,但這兩位本來可能夭折患兒都表現出雖然不完全但是顯著的持續的改善。
由德國Novartis Pharmaceuticals公司開展的一項臨床研究:抗硬化蛋白抗體BPS804 [3]用於治療HPP也已經報導了初步的短期結果。硬化蛋白是在稱為骨細胞的星形骨細胞中發現的蛋白質。硬化蛋白有助於減少或抑製(下調)被稱為成骨細胞的骨形成細胞。以往研究表明,抗硬化蛋白的抗體可增加骨質疏鬆症患者的骨量。


另外值得一提的一項進行中的研究是美國杜克大學 Durham 醫學中心正在做的一個前瞻性的研究[4],該研究旨在深入觀察HPP的臨床進展,如骨疾病的程度、眼科體征、整形外科問題、腎臟問題、肌肉骨骼表現,進而建立一個綜合性的多學科的模式來治療HPP,該研究從2014年開始,預計2026年能完成。
8
結語
目前人們所知的罕見病,80%以上是由遺傳變異所導致。而隨著基因測序和分析技術的飛速發展,人類基因組學、轉錄譜組學、蛋白質組學、代謝組學等研究的深入,基因檢測的簡單化、常規化、商業化,罕見病的診斷鑒別水準和發現率也有所增加,基因編輯技術的革命性突破所推動的罕見病動物模型的研發,這些都將有效促進罕見病分子機理的研究以及臨床前藥物研發的進展。未來相信會有更多孤兒葯被開發並從臨床試驗迅速進入真實世界應用,解決更多家庭與個人的痛苦。
參考文獻:
[1]Alexion (ALXN) Release: Long-Term Data Confirm Benefits Of Treatment With Strensiq (Asfotase Alfa) In Adolescents And Adults With Hypophosphatasia (HPP) Through Five Years
[2]MP Whyte,J Kurtzberg,WH Mcalister,S Mumm,MN Podgornik 《Journal of Bone & Mineral Research》,2003,18(4):624-636
[3] Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia
[4]Natural History Study of Patients With?Hypophosphatasia?(HPP) (NatHisHPP)
https://www.clinicaltrials.gov/ct2/show/NCT01406977?cond=hypophosphatasia&rank=5


罕見病專題
▼ 點擊閱讀原文,查看更多好內容