腫瘤細胞正在「勝利大逃亡」
去年備受關注的諾貝爾生理學或醫學獎,讓「免疫負調控」的發現者詹姆士·艾利森和本庶佑聲名顯赫。「免疫負調控」發生在抗原呈遞期間,即初始T細胞與抗原呈遞細胞之間傳遞抗原信號,以及細胞識別期間,即效應T細胞遷移進入腫瘤組織,與腫瘤細胞或免疫細胞之間傳遞識別信號期間。
大部分腫瘤細胞就是充分利用「免疫負調控」,來抑製細胞毒性T細胞的免疫活性,從而逃避免疫系統的追殺。具體地說,在正常狀態下,當炎症反應發生時,NK細胞、T細胞、巨噬細胞、樹突狀細胞等免疫細胞,以及表皮細胞和血管內皮細胞表面會被誘導表達PD-L1蛋白。當這些細胞和被激活的T細胞接觸時,PD-L1與T細胞表面的PD-1結合,從而抑製T細胞的免疫活性,避免過激的炎症反應對自身的傷害。因為這些細胞表面表達的PD-L1程度比較低,所以可以避免對T細胞活性的消耗。然而腫瘤細胞大不一樣,它們在細胞表面大量表達PD-L1,能夠幾乎完全抑製與它們接觸的所有T細胞的免疫活性,造成T細胞活性耗竭,並逃避免疫系統的追殺,最終惡性繁殖擴增,危及生命。(圖1)
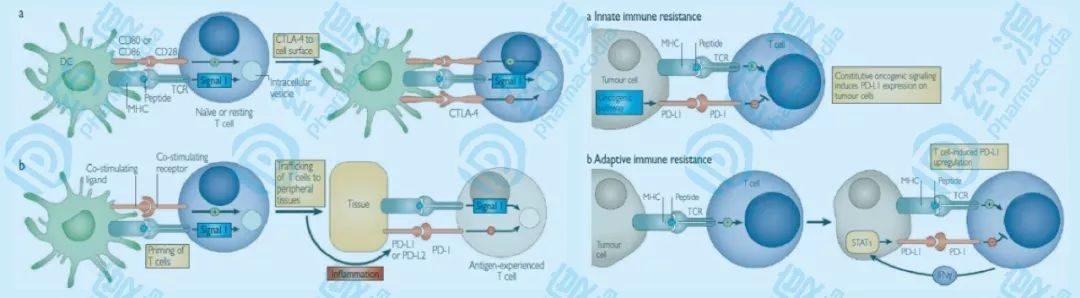
圖1 :免疫負調控示意圖
(a)當T細胞對抗原產生初次響應時,CTLA-4介導的免疫檢查點被誘導激活。這種由CTLA-4介導的誘導激活程度,依賴於起始T細胞受體調整的信號強度。高親和配體能夠誘導表達更多的CTLA-4,從而減弱了起始響應的強度。當T細胞受體遭遇抗原後,誘導下遊通路,CTLA-4被轉運到細胞表面,此時CTLA-4起到信號減弱的功能,以維持一個恆定的T細胞激活水準。(b)與CTLA-4不同,PD-1信號通路並不在起始T細胞激活階段起作用,而是在外周組織中,效應T細胞識別組織中的抗原,以調節炎症響應的過程中。這些組織中的炎症信號(IFN-γ,主要由I型輔助T細胞表達)能夠誘導組織細胞中的PD-L1的表達,從而抑製效應T細胞對其免疫響應。在慢性抗原暴露的情況下,T細胞表面過量誘導的PD-1,可以引發T細胞的活性耗竭。(c)在腫瘤細胞中,PD-L1的表達或不依賴於腫瘤微環境中的炎症信號,AKT、STAT3信號通路的激活可誘導表達PD-L1,或依賴於炎症信號,表達下遊的免疫檢查點抑製蛋白。
目前,腫瘤的聯合免疫療法越來越體現出它的優越性。一方面,要減少腫瘤靶向結合的非特異性,減少治療過程對正常細胞的殺傷,我們需要儘可能地將免疫系統引起的細胞毒性局限在腫瘤組織中(靶向藥物)。另一方面我們需要解除腫瘤細胞和免疫細胞之間的免疫負調控,提高免疫細胞對腫瘤細胞的細胞毒性(免疫負調控抑製),使腫瘤殺傷單抗藥物的腫瘤殺傷作用更能發揮威力。
在這些治療方案中,都以T細胞免疫為核心。雖然細胞毒性T細胞的腫瘤殺傷作用具有一定的特異性,然而極優而劣。因為細胞毒性T細胞的特異性,來源於被殺傷細胞的MHC-I型抗原遞呈。只有識別了目標細胞通過MHC-I遞呈的抗原,細胞毒性T細胞才能特異性的殺傷靶細胞。然而,狡猾的腫瘤細胞,有相當一部分關閉了其細胞表面MHC-I類分子的表達,比如,92%的宮頸癌細胞,71%的乳腺癌細胞,64%的非小細胞肺癌細胞。這樣我們英勇無比的細胞毒性T細胞,對它們就無能為力了。
「如之奈何?」
幸運的是,對付這類狡猾的腫瘤細胞,我們有免疫系統的另一殺手,自然殺傷細胞(Natural Killer Cell, NK細胞)。
我們日益信賴的替補選手: NK細胞
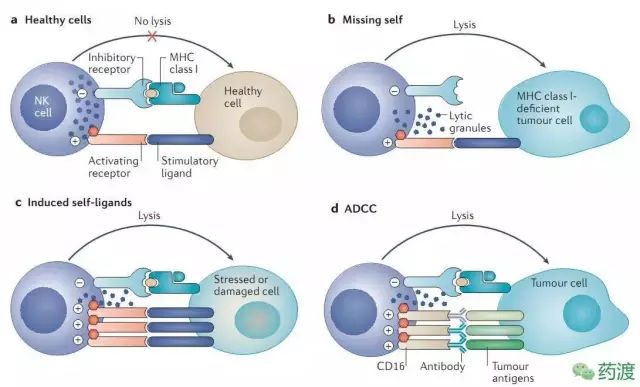
NK細胞,即自然殺傷細胞,是一種具有細胞毒性的淋巴細胞,屬於天然免疫系統。NK細胞與獲得性免疫系統中的細胞毒性T細胞,扮演著相近的角色。NK細胞對病毒感染的細胞,或者腫瘤形成,有著極快的響應速率。通常情況下,免疫細胞檢測到感染細胞表面的MHC,引起細胞因子的釋放,進而導致靶細胞裂解或凋亡。但NK細胞有所不同,它們可以在沒有抗體或MHC的情況下,識別這些細胞並進行快速的免疫響應。對於那些失去自身標記的MHC-I型的細胞,NK細胞不經過激活就可以進行殺傷。而這些細胞通常是有害的,不能被其他免疫細胞發現並消滅,比如細胞毒性T細胞。(圖2)

圖2 NK細胞的「丟失自我」的殺傷機制
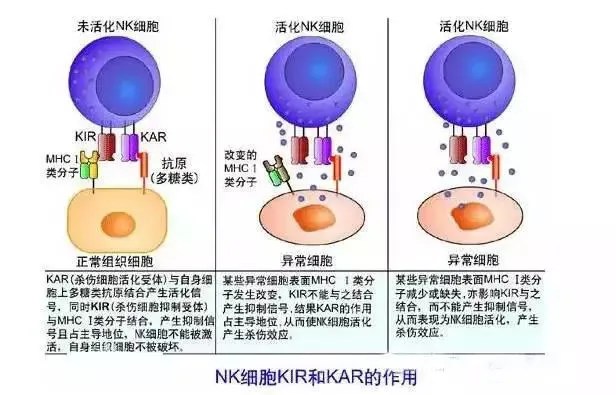
NK細胞通過自身表面的激活型和抑製型受體,調節自身的細胞毒性,比如殺傷細胞類免疫球蛋白受體。大部分受體不僅表達在NK細胞上,也表達在T細胞上。抑製型受體識別並結合MHC-I,這樣也可以解釋NK細胞殺傷那些沒有表達MHC-I的細胞。而MHC-I在抗原呈遞過程中,激活細胞毒性T細胞。但是,當被感染或變異的細胞,逐漸降低表達MHC-I,使它們自身免於被T細胞發現,規避T細胞免疫。而NK細胞正好彌補了這一點。
儘管NK細胞並不需要腫瘤相關抗原識別,來調整抗腫瘤的響應,但NK細胞同樣存在免疫檢查點的激活或抑製機制。雖然目前在細胞因子治療方法,以及NK細胞過繼轉移等方面有所進步,但是,腫瘤細胞表達的針對NK細胞免疫檢查點的配體,仍然能夠抑製NK細胞介導的腫瘤細胞裂解。於是NK細胞功能缺失,腫瘤逃逸,病情加劇。因此目前有一些新的藥物被研發出來,針對腫瘤-NK細胞的抑製型免疫檢查點,以限制這種抑製作用。
當細胞在病毒感染向腫瘤轉化期間,產生了應激壓力或DNA損傷,就會產生胚系編碼配體(germ-line ligand),這些配體可以被NK細胞表達的胚系編碼受體(germ-line receptor)識別。而當細胞出現低表達MHC-I的情況時,就會觸發「丟失自我」殺傷機制。因此為了最大程度地減少對正常細胞或組織的殺傷,必須微妙的平衡這種激活或抑製的信號,以調節NK細胞的活性。
NK細胞表面的激活或抑製型受體
NK細胞表面激活型的受體包括:自然細胞毒性引發受體(NCRs)、SLAM家族受體、c型凝集素、CD16(FcγRIII)。例如CD16並不識別細胞表達的配體,而是識別細胞結合的IgG抗體的Fc部分,而且單獨通過CD16就足夠引發強大的激活信號且克服大部分抑製信號,引發NK細胞-抗體介導的ADCC。另外,C型凝集素的同二聚體NKG2D,識別細胞表面因DNA損傷或應激壓力而上調的分子。這些激活型受體結合配體後,還能引發細胞因子比如IFN-γ、TNF-α的分泌,其中IFN-γ可誘導周圍細胞MHC-I的表達,增強CD8+ T細胞的識別能力(圖1)。
NK細胞表面抑製型的受體包括:殺傷細胞免疫球蛋白類似受體(KIRs)、c型凝集素受體(NKG2A/CD94)、白細胞免疫球蛋白類似受體(LILRs)、常見的免疫檢查點受體(PD-1、TIM-3、LAG-3、TIGIT)。它們當中大部分的配體,是MHC-I,而廣泛表達的MHC-I配體介導的抑製信號,對於NK細胞響應調節至關重要。這些抑製型受體的表達,因NK細胞亞群不同而不同,比如CD56bright的NK細胞都表達NKG2A/CD94,而不表達KIRs,但是CD56dim的細胞只有約50~60%表達NKG2A/CD94,70~75%表達KIRs。

圖3 NK細胞與腫瘤細胞間的激活或抑製型受體-配體相互作用
NK細胞的響應被這些激活或抑製型的相互作用的平衡微妙地調節,而這些NK細胞受體的表達取決於NK細胞的亞群,以及腫瘤微環境中的細胞因子或可溶性配體。同時,腫瘤細胞表達對應的配體,也依賴於腫瘤類型和微環境。
腫瘤縱橫諜海期間,一定程度地引起DNA損傷,這會誘導NKG2D和DNAM-1的表達,進而引發NK細胞對腫瘤細胞的殺傷。然而,腫瘤細胞可以通過上調非經典的MHC-I,即HLA-G的表達,結合NK細胞的抑製型受體LIR-1,規避NK細胞的識別和殺傷。同時,腫瘤細胞也可以通過可溶性的NKG2D的配體,規避殺傷。這些配體通過可變剪切的方式,從腫瘤細胞表面脫落。於是NK細胞難以通過激活型受體NKG2D與其配體結合,也就難以激活對腫瘤細胞的殺傷。同時,這些可溶性的NKG2D配體,可以結合遠處近處的NK細胞的NKG2D激活型受體,使其處於持續激活狀態,而降低了NK細胞識別的敏感性。
一些位於腫瘤微環境中的抑製型免疫細胞,比如骨髓衍生抑製細胞(MDSCs)、調節型T細胞(Treg)可以抑製NK細胞的抗腫瘤活性。MDSCs通過分泌抑製型細胞因子IL-10和TGF-β,其中TGF-β可以下調NK細胞表面NKG2D的表達,或者通過細胞接觸的方式,抑製NK細胞的活性。同樣的,Treg也可以通過膜表面的TGF-β抑製NK細胞的活性,且Treg也通過競爭性消耗IL-2以減少IL-2對NK細胞的激活。
「自然殺傷」替補選手的弱點
關於NK細胞的免疫檢查點,同樣存在著可能的負調控機制(圖3展示了一部分):PD-1,在B細胞和T細胞表面存在誘導性表達,同樣的,在NK細胞表面也有表達,儘管表達特性並不清楚,但PD-1減弱免疫功能的機制是明確的。不過,當NK細胞提升對腫瘤細胞的響應時,特別是IFN-γ分泌時,可能會導致腫瘤細胞PD-1配體的上調錶達,從而反饋抑製NK細胞的響應。
CTLA-4,在激活的鼠源NK細胞上發現存在表達。但目前幾乎沒有線索能直接說明,人源NK細胞表達CTLA-4的相關活性。
TIGIT,帶有Ig和ITIM(細胞內基於酪氨酸的抑製模體)結構域的T細胞免疫受體,通常在NK細胞上有所表達,屬於抑製型受體,與DNAM-1共享PVR和Nectin-2受體。許多腫瘤過表達TIGIT的配體,CD155,這與腫瘤的增殖和遷移有關。在腫瘤環境中,CD8+T細胞和Treg都會上調錶達TIGIT,而阻斷TIGIT能夠增強T細胞的功能。類似的,阻斷TIGIT能夠增強NK細胞分泌細胞因子和細胞毒性的能力。有數據表明,MDSC通過TIGIT信號通路,抑製NK細胞的活性。
KIR,殺手細胞免疫球類似受體,有抑製型和激活型兩種,針對抑製型的KIR的阻斷,是免疫治療的主攻方向。抑製型KIR有兩類,表達2個胞外免疫球類似結構域(KIR2DL),或者表達3個免疫球類似結構域(KIR3DL)。兩類KIR的信號通路都通過ITIM(細胞內基於酪氨酸的抑製模體)實現。KIR能夠識別並結合MHC-I,以抑製NK細胞的活性。在NK細胞的發育和穩態階段,KIR與自身的MHC-I的相互作用,對於NK細胞「教育」的動態過程非常關鍵。儘管在腫瘤環境中,NK細胞上調錶達激活型受體,但許多腫瘤能夠保留它們的MHC-I,從而能夠限制KIR表達NK細胞的響應和殺傷能力。而KIR信號通路的阻斷型抗體,能夠起到一定的腫瘤治療的效果(圖4)。
KIR抗體,IPH2101,在針對那些完全緩解狀態的急性髓細胞樣白血病患者的I期臨床研究中,顯示KIR結合發生在90%以上的NK細胞中(2周,最小劑量1mg/kg體重)。KIR抗體的治療也升高了TNF-α和MIP-1β的血清濃度,以及NK細胞早期的激活標籤CD69。在多發性骨髓瘤的治療中,KIR抗體也產生了類似的效果。然而,在關於鬱積性多發性骨髓瘤(MM)的II期臨床中,並沒有顯著療效。這可能與IPH2101介導的KIR2D受體的胞啃作用有關,即通過KIR抗體的ADCC作用,KIR2D受體被「啃掉」並轉移到其他免疫細胞。儘管KIR抗體有效地阻斷了KIR2D的信號通路,但是也阻斷了這些表達KIR2D的NK細胞被「教育」(結合MHC-I)的能力,最終可能導致NK細胞對MM細胞的響應清零。這個難題也揭示了,在複雜的生物系統中,靶向檢查點抑製研發所存在的一些挑戰。
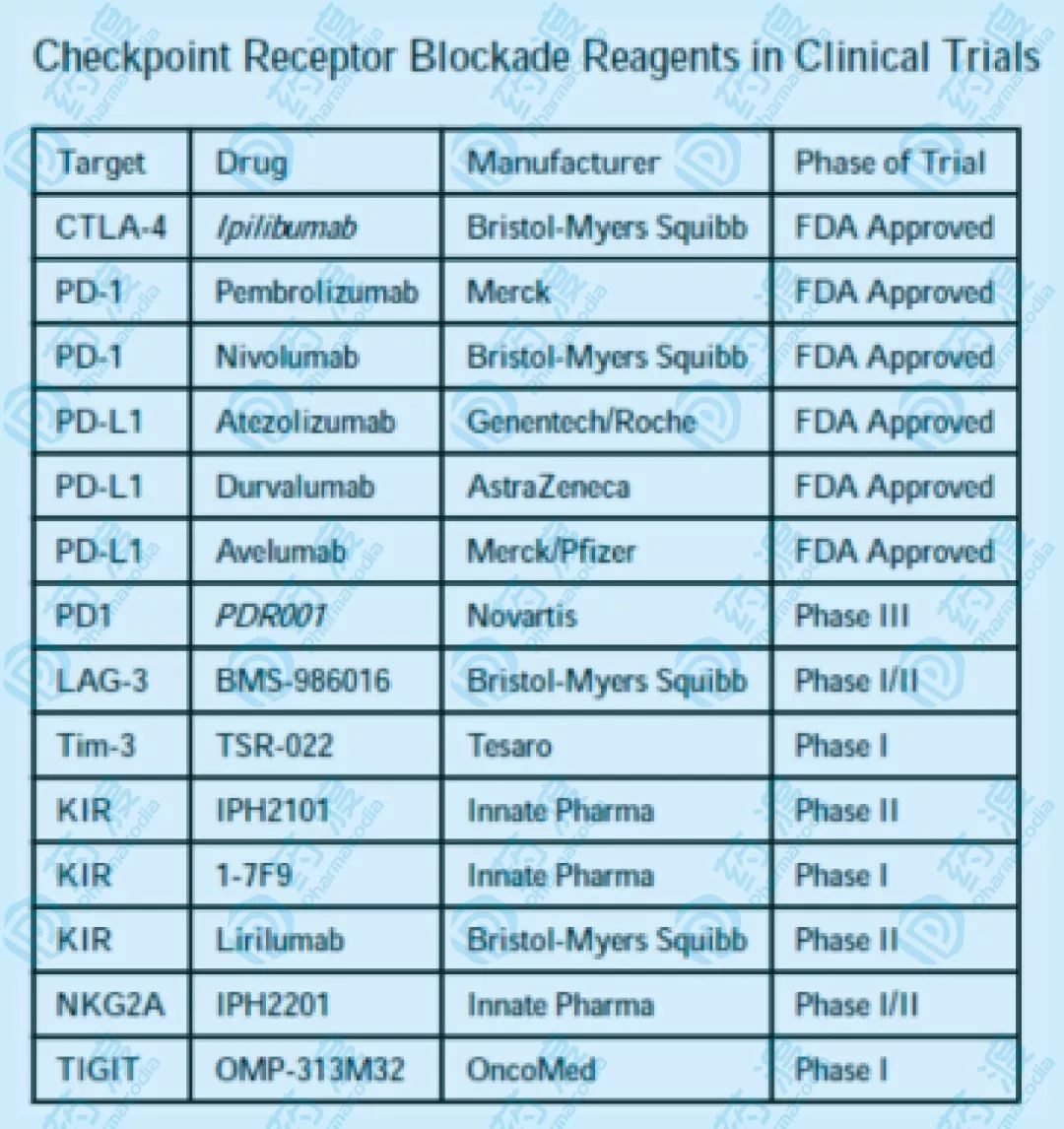
圖4 NK細胞相關免疫檢查點的抗體,以及它們的臨床研究進展(截至2017年)
C型凝集素異二聚體NKG2A/CD94,在NK細胞和CD8+T細胞上都有表達。它屬於抑製型受體,對應的配體是HLA-E,可強烈抑製血液循環中的NK細胞。在很多腫瘤類型比如實體瘤或血癌中,上調編導HLA-E,從而減弱表達NKG2A的NK細胞的響應。在異源和自體造血乾細胞移植中,NKG2A廣泛表達於新生的NK細胞上,它與HLA-E的相互作用成為移植性治療後NK細胞活性的主要抑製因素。在這個條件下,NK細胞通過減少NKG2A的表達,恢復NK細胞功能,並最終成熟。但在NK細胞完全成熟之前,阻斷NKG2A也可以恢復功能,因此NKG2A的功能抑製型抗體也有希望用於治療腫瘤。
Tim-3,T細胞免疫球和黏液素結構域包含分子,是T細胞調節免疫響應的負調節因子。小鼠中抗Tim-3的作用,導致自發性的自身免疫作用。在晚期胃癌和肺腺癌患者的外周NK細胞中,Tim-3上調錶達。同時,在75%的胃腸道間質瘤患者中,腫瘤濾過性的NK細胞上有表達Tim-3。NK細胞表達的Tim-3的具體功能並不明確。源自晚期黑色素瘤的患者的功能受限的NK細胞,通過Tim-3拮抗劑的治療,功能可獲得恢復。然而,阻斷Tim-3和它的配體galectin-9的相互作用,減少了健康NK細胞對急性髓性白血病(AML)的IFN-γ的產生。在對PD-1的阻斷有抗性的患者中,Tim-3發生了阻斷,同時Tim-3表達被上調,因此Tim-3被認為存在抑製免疫活性的作用。
Lag-3,淋巴細胞激活基因3,表達在CD4+和CD8+T細胞上。因只有小部分NK細胞表達Lag-3,並且NK細胞與MHC-II並不相互作用,因此Lag-3的功能還不是很清楚。
NK細胞的神助攻:激活型細胞因子

圖5 IL-2/IL-2Rα和IL-15/IL-15Rα的結構示意圖
IL-2和IL-15共享受體的兩個亞基,即IL-2Rβγ和IL-15Rβγ完全相同,雙亞基複合體以中親和力受體存在,而結合了IL-2Rα或IL-15Rα的三亞基複合體,則以高親和力受體存在。NK細胞表達中等親和力的IL-2和IL-15受體。通常情況下,IL-2通過順式作用,即先於同一細胞的IL-2Rα結合,然後與βγ結合,但NK細胞沒有表達IL-2Rα,需要較高濃度的IL-2才能激活。而IL-15通過反式作用,即先於另一細胞(巨噬細胞、DC細胞)的IL-15Rα結合,然後與βγ結合。
NK細胞可以通過兩種方式克服抑製,一是激活型細胞因子,比如IL-2、IL-15,二是CD16(FcγRIII)介導的NK細胞激活。IL-2和IL-15共享相同的βγ-受體亞基,而各自的α受體亞基可以提高它們在受體上的親和力,它們結合對應的高親和或中親和受體,激活JAK-STAT信號通路,最終誘導更多的細胞因子的表達,細胞毒性的效應功能,以及增殖和存活。
IL-2治療已經被大量研究,但是單獨使用IL-2所產生的療效並不顯著。IL-2的一項缺點是,儘管它能夠激活NK細胞,但同時,它也可以增強Treg的活性,限制NK細胞的響應。高劑量IL-2治療腎癌或轉移性黑色素瘤,只能讓小部分患者的病情緩和,並且殘留有大量細胞毒性。因為低劑量的IL-2更利於激活Treg的功能,所以IL-2治療受限,而IL-15對於NK細胞依然有較好的刺激效果,且不會激活Treg的功能。
IL-15被用於實體瘤的治療,以及在白血病患者中,維持NK細胞數量和活性。臨床前、非人類哺乳動物以及早期臨床的數據都表明,IL-15可以誘導NK細胞數量上升。其中IL-15與IL-15Rα的反式呈遞,對最大化IL-15功效是必要的。ALT-803(IL-15N72D/IL-15Rα-Fc超級激動劑),在最近針對卵巢癌的鼠模型研究中,使NK細胞非常顯著地去顆粒化,並導致細胞因子的大量產生。如ALT803能夠使卵巢癌患者腹水中的NK細胞恢復活性。同時,在抗CD20抗體的聯合作用下,ALT803增強了CD16引發NK細胞對B淋巴細胞的清除。IL-15一方面能夠協助越過免疫檢查點的抑製機制,另一方面能夠完善NK細胞上CD16介導的功能,因此它成為了新的熱門免疫治療靶點。
CD16a,即FcγRIIIa,NK細胞表達的Fc低親和受體,介導抗體的直接殺傷ADCC。CD16a表達在CD56dim的NK細胞上,這些NK細胞在健康個體內,佔有至少80%的所有外周的NK細胞。CD16a與Fc結合後,通過ITAM信號通路,導致細胞因子的產生和細胞的去顆粒化。與NK細胞的其他激活型受體不同,CD16a與Fc的結合不需要協同激活,就可以產生強力的響應,這也使得NK細胞能夠在病毒感染和腫瘤形成早期,通過抗體產生免疫反應。然而,NK細胞表達的CD16a對不同抗體的Fc親和力有所差異,介導的ADCC的效果參差不齊。因此,開發BiKE和TriKE之類的分子,能改進親和力,並且可以針對不同的腫瘤相關抗原。

圖6 BiTE和BiKE
BiTE能夠同時結合T細胞和腫瘤細胞,具有兩條scFv串聯結構的抗體,分別結合T細胞的CD3ε(TCR亞基)和腫瘤細胞的腫瘤相關抗原。而BiKE則能夠同時結合NK細胞和腫瘤細胞,結構與BiTE相近,結合NK細胞的CD16a,強化ADCC作用,以及腫瘤相關抗原。新一代的BiTE甚至加入了抗PD-1/PD-L1的scFv,以減少免疫檢查點抑製。類似的原理(圖3),BiKE可以融合IL-15或拮抗前述受體信號通路的scFv(TriKE),移除免疫抑製或增強免疫響應。
腦洞大開的NK細胞腫瘤免疫治療
激活NK細胞、解除NK細胞的免疫抑製、限制NK細胞毒性的時空。
新的針對NK細胞的研究,比如仿照BiTE思路而來的BiKE(圖6),以及仿照CAR-T思路的CAR-NK(另文描述)。BiTE加上抗PD-1的部分成為了CiTE(檢查點抑製T細胞連接抗體),那麼BiKE加上了IL-15的部分而成為了TriKE(三功能NK細胞連接抗體,圖7,8):

圖7 CiTE和TriKE

圖8 TriKE的臨床研究正在招募中
如果說,TriKE給我們提供了增強NK細胞的免疫響應和特異性的參考,那麼如何武裝NK細胞,是可以關注的點。一方面,抑製NK細胞的抑製性信號通路,比如抗體阻斷NK細胞表面的抑製型受體,或者激活NK細胞,通過結合激活型受體的配體,比如IL-2、IL-15、Fc等;另一方面,提高NK細胞,尤其是滲透到腫瘤組織的NK細胞的有效性,提高針對腫瘤細胞的特異性,比如利用抗腫瘤的單抗。那又如何將這幾方面的優勢綜合起來「武裝」NK細胞?
細胞因子對腫瘤的治療效果,因為受到藥物傳遞的限制,無法在腫瘤病灶部位產生足夠的活性,比如之前提到過IL-2的局限性。為了最大化細胞因子的治療效果,重組的抗體-細胞因子融合蛋白被廣泛研究,以增強單抗靶向腫瘤的能力。如此,細胞因子通過單抗,被引導至特異性的腫瘤部位,能夠刺激引發更多有效的抗腫瘤響應,並且避免單獨使用細胞因子時會造成的系統性的細胞毒性。

圖9 用於免疫治療的細胞因子,其中一些還處於臨床研究階段
細胞因子是多肽或蛋白質,在重組基因表達上,容易實現在哺乳動物細胞中的表達,只需將INFs、ILs之類的融合在抗體的N端或C端。比如羅氏在研的RG7813(圖10),包含抗CEA單抗,且其中一條重鏈的C端融合改造後的IL-2。同樣的,前文提到的TriKE,基於scFv抗體,以拉近NK細胞和腫瘤細胞,並通過融合細胞因子激活NK細胞的活性。

圖10 RG7813示意圖
抗CEA單抗特異性結合CEA最靠近細胞膜表面的結構域,而不結合因酶切作用而進入血液的可溶性部分,sCEA屬於腫瘤細胞抑製體液免疫的作用。IL-2通過突變改造,偏向於結合IL-2中等親和力受體IL-2Rβγ,從而激活NK細胞或CD8+細胞毒性T細胞。因為IL-2存在,Fc的ADCC/CDC的活性被取消,為了長效作用仍保持FcRn的結合,並且因為單側結合IL-2,引入KiH技術實現Fc的異二聚化。
設計理想中的抗體-細胞因子融合蛋白,包括以下步驟:
選擇合適的靶向抗原:在正常組織中表達量極低的腫瘤相關抗原,以減少抗體「沉沒」在正常組織中而無法發揮藥效,同時也需要腫瘤組織中儘可能高表達,以便富集足夠多的細胞因子。細胞表面的抗原,結合後不會被細胞吸收,這樣可以延長融合細胞因子的半衰期。腫瘤微環境相關抗原也可以,比如與腫瘤遷移、浸潤相關的細胞因子。在血液循環中沒有顯著的數量,比如sCEA,能夠持續性消耗激活的免疫細胞,使這些免疫細胞無法造成有效殺傷。
選擇合適的細胞因子:考慮細胞因子靶向到腫瘤細胞的目的,還沒有一種單一的細胞因子具有所有的抗腫瘤的特性。是為了激活和輔助增殖T細胞、NK細胞還是巨噬細胞?是否需要直接作用於腫瘤細胞,還是用於抑製腫瘤組織血管的形成?細胞因子介導的殺傷機制,誘導T細胞的細胞毒性,還是ADCC,還是直接作用?本文優先考慮對NK細胞的激活作用。
結構的選擇:完整的抗體結構,scFv,Fab等等。
融合細胞因子的生物活性:通過定量比較,維持完全的親和力和生物活性,或者減弱活性以減少脫靶造成的系統性細胞毒性。正常組織中,過高的親和力可能導致細胞毒性。而結合到靶細胞後,也要注意抗體介導的巨噬細胞吞噬,除非細胞因子靶向作用於巨噬細胞。這會使融合的細胞因子被巨噬細胞吞噬和消除。因此,如果用到了Fc融合,Fc往往需要通過改造以減少細胞因子的非特異消耗。
PK和PD:足夠長的半衰期,以便於在腫瘤部位富集。
臨床前藥效估計:在腫瘤模型動物實驗中,檢測抗腫瘤藥效和免疫毒性。
劑量和使用規劃:在腫瘤微環境以及宿主的免疫效應細胞的條件下,基於耐受性和葯代的效果制定。
結 尾
抗體細胞因子的融合,要求它針對腫瘤細胞的活性和有效性,強於單獨用抗體或單獨用細胞因子的效果,否則聯合用藥可以達成更好的效果。因此,最好在相關同基因的免疫活性動物模型中,驗證融合蛋白的效果。
最後,以TriKE的構建理念為結尾,一方面靶向腫瘤細胞,一方面靶向NK細胞,激活NK細胞或者解除NK細胞的免疫抑製,然後通過IL-2或IL-15的改造體激活NK細胞的活性。如果融合的細胞因子,在TriKE功能的基礎上,活性可限制在腫瘤微環境中釋放,而且本身在血液循環中有較長的半衰期,這可能比TriKE更為理想。
參考文獻
1. NLRC5/MHC class I transactivator is a target for immune evasion in cancer.(2016), PNAS, Sayuri Yoshihama, Jason Roszik, Isaac Downs, et al.
2. Trends in the global immuno-oncology landscape. (2018) Nature Reviews. Jun Tang, Laura Pearce, Jill O』Donnell-Tormey and Vanessa M.Hubbard-Lucey.
3. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. (2018) Cell. Miguel F. Sanmamed and Lieping Chen.
4. The basic principles of chimeric antigen receptor (CAR) design. (2013) Cancer discovery. Michel Sadelain, Renier Brentjens, and Isabelle Riviere
5. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. (2017) Oncoimmunology.Christian Klein, Inja Waldhauer, et al.
6. Targeted killing of colorectal cancer cell lines by a humanised IgG1 monoclonal antibody that binds to membrane-bound carcinoembryonic antigen.(2008) British Journal of Cancer.PJ Conaghan, SQ Ashraf.
7. Natural Killer Cells Unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. (2017) SeminImmunology. Zachary B. Davis, Daniel A. Vallera, Jeffrey S. Miller, and Martin Felices.
8. Antibody-cytokine Fusion Proteins for Treatment of Cancer: Engineering Cytokines for Improved Efficacy and Safety. (2014) Semin Immunology. PatriciaA. Young, Sherie L. Morrison, and John M. Timmerman.
9. ESMO HANDBOOK OF IMMUNO-ONCOLOGY. Edited by John B.A.G. (2018) Haanen,Raffaele Califano, Iwona Lugowska, Marina Chiara Garassino.