▎葯明康德/報導
編者按:罕見病不罕見。根據世界衛生組織的統計,全球約有5000-8000種罕見病。儘管單一疾病的患者人數不多,但當這些患者聚在一起,總人數將接近4億。也就是說,每不到20個人裡,就有一名罕見病患者。在今年的國際罕見病日(Rare Disease Day),葯明康德微信團隊希望通過這篇故事,為全球4億名罕見病患者及他們的家庭帶來生活的信念與堅持的勇氣。
馬特·威爾西(Matt Wilsey)的生活令人羨慕。他出生於舊金山一個頗有名望的家族,在優越的家境中接受了極好的教育。在史丹佛大學順利畢業後,馬特在矽谷盡情揮灑才華,打造了多家價值數千萬美元的科技新銳。
事業有成的馬特,感情生活也同樣美滿。2009年,他的妻子克麗絲滕(Kristen Wilsey)懷孕了。兩人給孩子起了格瑞絲(Grace)的名字,期待著愛情結晶的誕生。沒有人會想到,他們等來的,竟是人生的急轉彎。
突如其來的命運
2009年10月24日,意外悄然而至。還在母親肚子裡的格瑞絲心率突然下降,情況一度非常危急。克麗絲滕被緊急送往史丹佛大學醫院接受剖腹產。儘管手術很順利,但馬特卻隱隱感到心神不寧。他屢次把醫生叫來檢查格瑞絲,而醫生的回答總是格瑞絲沒有問題,請他放心。
時間的推移非但沒有打消馬特的擔憂,反而讓他的恐慌與日俱增。剛出生的格瑞絲目光渙散,不願進食。幾天后,她的肝功能又出現了問題,在新生兒重症監護病房待了足足兩周。令人不安的是,醫生做足了檢查,也不知道她究竟出了什麼問題。


▲克麗絲滕,格瑞絲,以及馬特(圖片來源:Grace Science Foundation為葯明康德特別提供)
脫離危險期後,格瑞絲被無奈的醫生要求回家靜養觀察。然而隨著時間推移,格瑞絲的病情愈發嚴重起來,還出現了許多令人擔憂的癥狀:與同齡人相比,她的認知能力與運動能力發育得太慢了;肌張力低下讓她全身癱軟,就好像是個布偶娃娃;而從出生起就困擾她的肝功能問題,也絲毫沒有得到減退。
本文來源:葯明康德
與所有關愛子女的父母一樣,馬特與克麗絲滕盡自己全力為女兒尋找病因。他們從美國的西海岸尋找到東海岸,造訪了能想到的所有頂尖醫院,諮詢了100多位醫生。儘管出現的癥狀越來越多,諮詢的醫生越來越資深,格瑞絲依舊沒有得到一個確切的診斷。
這並不能責怪醫生們。要知道,人類首次記載這種疾病,還是幾年後的事情。當時,它處於人類的未知領域,沒有人知道它的存在。
生命藍圖的錯誤
2003年,人類基因組計劃宣告完成。這一里程碑不僅開啟了生物學的新紀元,還大大降低了發現基因突變的難度。如果一名患者的罕見病情是由基因突變所引起,我們就有機會通過「基因組測序」,找到遺傳密碼裡的病因。
我們的基因組裡,繪製著生命的藍圖。然而基因突變就像是印刷錯誤,難以避免。大多數情況下,這些錯誤帶來的影響有限,無傷大雅。極罕見的情況下,這些突變會造成嚴重後果,危及生命。
由於格瑞絲的病情遲遲無法得到診斷,許多醫生懷疑她是不是得上了某種還不為人知的罕見病。於是在兩歲那年,格瑞絲的父母帶她前往多家頂級醫學機構進行全基因組測序,尋找答案。在著名的貝勒醫學院,困擾他們多年的難題終於得到了解答。


▲馬修·貝恩布裡奇找到了問題的答案(圖片來源:馬修個人LinkedIn主頁)
答出這道難題的是當時還在攻讀博士學位的馬修·貝恩布裡奇(Matthew Bainbridge)。拿到格瑞絲的基因組測序數據後,馬修先在其中尋找了一遍已知的致病基因突變,卻沒有搜索到任何結果。
隨後,他又在其中尋找那些先前沒有被科學家們發現,卻可能導致疾病的突變。這一次,他找到了突破口。馬修發現,格瑞絲的兩條NGLY1基因(一條來自父親,一條來自母親)都出現了嚴重的突變。用他的話說,「一處突變非常糟糕,另一處突變也挺糟糕的。」由於這些突變,格瑞絲出現了NGLY1缺陷症。
不會哭的孩子
從基因序列上看,NGLY1編碼了一種叫做N-聚醣酶1的酶,它能從錯誤摺疊的蛋白質上移除糖分子,有助於細胞降解這些錯誤蛋白。當時,人們並不知道它怎樣導致了格瑞絲的癥狀。或許,NGLY1基因上的突變讓她的細胞積累了太多「垃圾蛋白」,損害了她的健康。
但馬修知道他找對了方向。隨後,他投入了浩瀚的資料中,尋找關於NGLY1基因的更多資料。幸運的是,他很快就找到了關鍵線索——2012年,杜克大學的科學家們發表了一篇論文,探討「全外顯子測序」能如何在臨床上加速罕見病診斷。研究裡,12名無法順利得到診斷的罕見病兒童以及他們的父母通過外顯子測序,找到了可能導致疾病的基因突變。其中,一名小男孩的體內檢測到了NGLY1突變。


▲人類首次報導NGLY1缺乏症,是格瑞絲出生3年後的事了(圖片來源:參考資料[5])
這名小男孩叫做伯特蘭。巧合的是,他的父親也叫馬特(Matt Might),當時是猶他大學的一名助理教授。「我的兒子伯特蘭得了一種全新的遺傳病,」他在部落格上寫道:「我的兒子是已知唯一缺乏這種酶的人類。」
葯明康德相關閱讀:當你是世間唯一的病人:致全球4億名罕見病患者
在部落格上,伯特蘭的父親詳細地記錄了兒子的病情,它們與格瑞絲的癥狀很像。馬修翻遍了這些博文,發現了一個很罕見的現象——伯特蘭不會哭。
格瑞絲會哭嗎?馬修很快發去了一封郵件。回信中,馬修得到了他想要的答案:據格瑞絲的母親回憶,3年多裡,她只見過格瑞絲哭過一兩次。難受的時候,格瑞絲的眼睛裡會有些濕潤,但幾乎從不掉淚。
這是屬於馬修的「尤裡卡時刻」。兩名患者幾乎一致的突變和癥狀,讓他終於能夠確認,NGLY1基因突變就是格瑞絲的病因。
一名父親的戰鬥
確診病因對這兩個家庭而言都是一個解脫,這意味著在黑暗中經過了漫長而痛苦的診斷探索,他們終於有了一個答案。但這個答案無助於兩名罹患罕見病的兒童告別疾病。2013年,伯特蘭6歲,格瑞絲3歲,他們無葯可用。
這是許多罕見病家庭所面臨的無奈。一年多前,還無人知曉這種疾病的存在;資源有限,很少有醫藥公司願意投入生物學機制不明的罕見病新葯研發;即便有公司願意投入其中,一款新葯從研發到上市,平均也需要約10年的時間,成功率僅為10%。
血管中流淌著創業者精神的馬特很快就找到了解決方案。既然沒有人在研發治療NGLY1缺陷的藥物,他決定親自上陣。2014年,以女兒的名字,馬特創立了一個科學基金會。在那裡,對生命科學一竅不通的馬特募集了數百萬美元的科研資金,並開始聯繫來自大學、研究所、以及生物技術公司等各個領域的科學家,希望他們能夠參與治療NGLY1缺陷的研究中。


在生命面前,素昧平生的陌生人,也可以成為志同道合的合作夥伴。受馬特的熱情所鼓舞,100多位頂尖科學家決定助他一臂之力,其中就包括了諾貝爾生理學或醫學獎得主山中伸彌教授。這些科學家組成了一個結構鬆散,卻目標明確的團隊,從不同角度探索NGLY1致病的生物學機制。


▲諾獎得主山中伸彌教授(前排左二)也向馬特伸出了援手(圖片來源:Grace Science Foundation為葯明康德特別提供)
在格瑞絲確診時,全世界已知患有NGLY1缺陷症的患者只有不到5位,這對於科學研究來說,樣本太小了。為此,馬特又與其他病患家庭合作,在全球尋找更多患有NGLY1缺陷的病患。互聯網時代,他們的聲音傳遍了世界的每一個角落。在他們的努力下,又有數十個患者家庭浮出水面。如今,確診的患者總數已經接近50,增長了10倍。


▲馬特造訪香港的一個罕見病家庭,男孩Tai Tai也是全球數十位NGLY1缺乏症患者之一(圖片來源:Grace Science Foundation為葯明康德特別提供)
在基金會的組織下,這些分散在全球的罕見病家庭於前年齊聚加州,為研究者提供了寶貴的血液和組織樣本。更重要的是,面對面的交流讓他們知道自己並不孤單,更讓他們看到有多少科學家正在為他們的子女奮鬥。這樣的心靈慰藉,是無價的珍寶。
與時間的賽跑
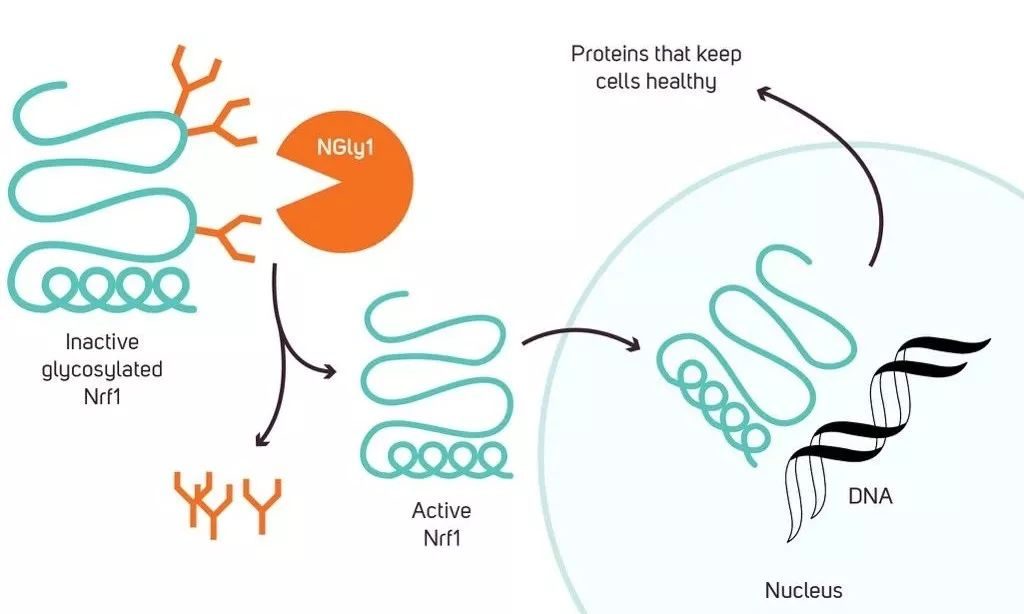
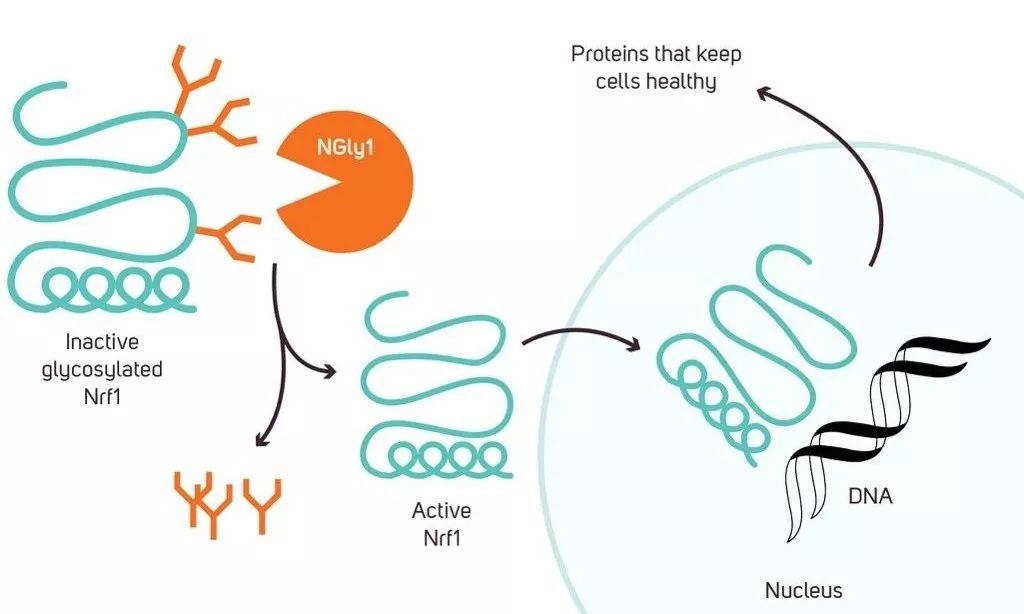
因為相信,所以看見。2016年,沒有放棄希望的馬特終於等來了曙光。作為協助馬特的100多名科學家之一,麥克阿瑟天才獎得主卡羅琳·貝爾托齊(Carolyn Bertozzi)教授精準地揭示了NGLY1蛋白參與的生物學通路——在細胞內,有一種叫做Nrf1的轉錄因子。只有當它表面的糖分子被移除後,才能發揮正常作用。而NGLY1的功能,正在於此。


▲麥克阿瑟天才獎得主卡羅琳·貝爾托齊教授揭示了NGLY1的生物學通路(圖片來源:Kuebi = Armin Kübelbeck [CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)])
倘若患者體內缺乏NGLY1蛋白,Nrf1就無法正常工作,這會影響蛋白酶體相關基因的表達,最終影響到細胞內的蛋白降解。於是,「垃圾蛋白」在細胞裡越積越多,導致疾病。這個發現證實了人們之前的猜測。
更重要的是,這條通路中的蛋白酶體,參與到了許多重要的疾病之中,這包括了癌症和大腦疾病。從罕見病研究出發,我們收穫的新知,有望帶來更深遠的影響。
馬特知道,是時候邁出下一步了。2017年,他募集了700萬美元的資金,正式成立了一家生物技術公司,探索NGLY1在治療多種人類疾病中的臨床潛力。和他的基金會一樣,這家公司以格瑞絲的名字命名。
在生物醫藥行業,這樣的例子並不罕見。1991年,一名德州男孩被診斷患有罕見的1型黏多糖病。在他的父母募集的研究資金下,科學家和醫藥公司帶來了首款治療這種罕見病的新葯。葯明康德集團合作夥伴Amicus的首席執行官約翰·克勞利(John Crowley)也是因為想要治療女兒的龐貝氏症,才創立了這家公司。
當然,新葯研發從來不是一件容易的事。馬特的許多朋友也都給他打了預防針。但對於馬特來說,他沒有其他選擇。目前,罹患NGLY1缺乏症的患者,年紀最大的也只有20出頭,更多會在成年前夭折。而在今年,格瑞絲已經10歲了。


▲格瑞絲今年已經10歲了。找到治療她的新葯,是一場與時間的賽跑(圖片來源:Grace Science Foundation為葯明康德特別提供)
馬特坦言,他不知道自己的女兒還能活多久。但在那一刻到來之前,他將繼續與時間賽跑。
參考資料:
[1] Grace Science Foundation official website, Retrieved Rebruary 26, 2019, from https://gracescience.org
[2] This Father Founded A Medical Research Startup To Save His Kid』s Life, Retrieved February 25, 2019, from https://www.fastcompany.com/40490486/this-father-founded-a-medical-research-startup-to-save-his-kids-life
[3] A FAMILY』S RACE TO CURE A DAUGHTER』S GENETIC DISEASE, Retrieved February 25, 2019, from https://www.wired.com/story/a-familys-race-to-cure-a-daughters-genetic-disease/
[4] Kids who don"t cry: New genetic disorder discovered, Retrieved February 25, 2019, from https://edition.cnn.com/2014/03/20/health/ngly1-genetic-disorder/index.html?no-st=1551082731
[5] Anna Need et al., (2012), Clinical application of exome sequencing in undiagnosed genetic conditions, DOI: http://dx.doi.org/10.1136/jmedgenet-2012-100819
[6] NGLY1 gene, Retrieved February 27, 2019, from https://ghr.nlm.nih.gov/gene/NGLY1
[7] Carolyn Bertozzi, (2017), A Model for Accelerating Patient-to-Bench Research, ACS Central Science, DOI: 10.1021/acscentsci.7b00540
[8] Frederick M. Tomlin et al., (2017), Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity, ACS Central Science, DOI: 10.1021/acscentsci.7b00224
TAG: |