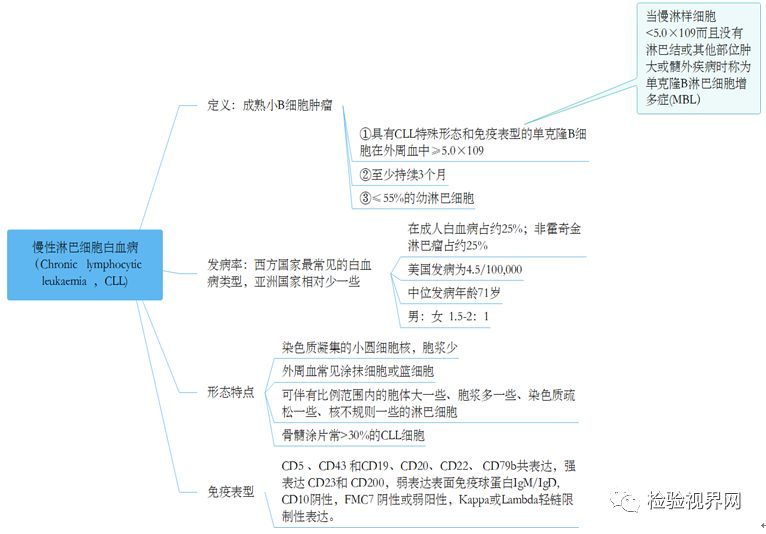
慢性淋巴細胞白血病(CLL)是一種以成熟B淋巴細胞在外周血、骨髓和淋巴組織蓄積,併產生相應臨床癥狀的B細胞慢性淋巴增殖性疾病(B-CLPD)。筆者在本文主要探討如何診治CLL以及診治過程中應該注意的一些問題,供感興趣的同行借鑒。
關於CLL診斷
例1,男,60歲,因"活動後心悸、氣短、乏力2個月"於2011年12月入住外院。體格檢查無淺表淋巴結腫大和肝脾腫大。血常規:WBC 378.11×109/L,RBC 1.77×1012/L,HGB 56 g/L,紅細胞平均體積(MCV)108.5 fl,紅細胞平均血紅蛋白(MCH)31.6 pg,紅細胞平均血紅蛋白濃度(MCHC)292 g/L,PLT 107×109/L,網織紅細胞0.005,中性粒細胞0.04,小淋巴細胞0.92,幼稚淋巴細胞0.04(在淋巴細胞中佔4.2%)。骨髓象:增生極度活躍,粒系及紅系增生減低,淋巴細胞比例明顯增高,幼稚淋巴細胞0.080。流式細胞術免疫分型:CD19+、CD5+、CD23dim(dim:弱表達)、FMC-7-、CD22dim、CD20bright(bright:強表達)、sIgλ+(中等強度表達)。常規核型分析未見分裂象。FISH檢測示:IGH-CCND1[t(11;14)(q13;q32)]陰性。診斷為CLL。給予FCR(氟達拉濱、環磷醯胺、利妥昔單抗)方案治療6個療程達完全緩解(CR),6個月後複發入住我院。流式細胞術示免疫表型檢測同上,且CD200 dim、CD148bright,骨髓免疫組織化學(IHC)檢查示:CCND1-、SOX11+、LEF1-。採用IGH-CCND1探針行FISH檢測,示融合基因陰性、IGH信號分裂,提示存在IGH基因易位(但夥伴基因不明)。CpG刺激的外周血染色體核型分析示:47,XY,t(12;14)(p13;q32),+12[3]/47,idem,del(9)(p21)[3]。免疫球蛋白重鏈可變區(IGHV)基因無突變。診斷為套細胞淋巴瘤(MCL)。本例患者是誤診為CLL的MCL。患者初診時外周血塗片以成熟小淋巴細胞為主,外周血B淋巴細胞≥5×109/L,且流式細胞術免疫分型確認其克隆性。CLL免疫表型積分為4分(CD5+、CD23+、FMC-及CD22dim各1分),診斷CLL似乎不存在問題。但CLL的典型免疫表型應為:CD19+、CD5+、CD23bright、sIgdim、CD20 dim、CD22 dim、FCM7-/dim,該患者有的表型(如CD20bright、sIg+中等強度表達、CD23dim)不是CLL的典型表型,所以必須與其他B-CLPD相鑒別。首先需要鑒別的是MCL,如FISH檢出t(11;14)陽性,則可以診斷MCL,以往我們據此診斷了多例與本例免疫表型類似的MCL,而此例患者未檢出t(11;14),我們認為應該診斷為CLL。複發時CD200dim也支持CLL的診斷,但CD148bright明顯高於CD200dim支持MCL,且骨髓IHC顯示LEF1-,不支持CLL(LEF1+為CLL/SLL特徵性標誌,其他B-CLPD則LEF1-),而約25%的MCL可CD23dim[3]、少數可表達CD200,關鍵是SOX11+,提示患者為少見的CCND1- MCL,染色體為t(12;14)(p13;q32)[12p13為CCND2基因所在部位],提示為罕見IGH-CCND2 MCL(累及CCND2或CCND3者,夥伴基因為免疫球蛋白輕鏈更常見)。由於成熟B細胞增殖能力低,初診常規核型分析未見分裂象,而可能如CLL,複發時採用CpG刺激可增加異常核型的檢出率。另外,需要強調的是,對於CLL等B-CLPD,外周血是進行塗片分類、染色體核型分析、分子生物學檢查的最佳標本(較骨髓好)。綜上,對於免疫表型除需注意是否陽性表達外,表達強度在診斷中也應引起足夠關注。對於部分免疫表型不典型的CLL,比如CD23-/dim、sIgbright、CD20bright等,建議行IHC檢測Cyclin D1和SOX11表達,同時行FISH檢測t(11;14),以排除MCL。LEF1、CD200、CD148等新的指標在CLL、MCL的鑒別診斷中也具有重要意義。不典型的CLL,如淺表淋巴結腫大等可以獲得病理標本的患者,應盡量行病理活檢。
關於CLL治療指征
例2,男,68歲。因"體檢發現白細胞增多1個月"就診。患者1個月前體檢,血常規:WBC 34×109/L,淋巴細胞0.86,HGB 131 g/L,PLT 254×109/L。查體未見淺表淋巴結腫大和肝脾腫大。外周血塗片細胞形態學檢查:可見大量成熟淋巴細胞聚集伴有較多塗抹細胞。生化檢測肝腎功能、LDH、β22-MG)正常。流式細胞術檢測示:可見單克隆B細胞,CD5+、CD19+、CD23+、CD200+、CD20 dim、CD22-、FMC-7-。CpG刺激的外周血染色體核型分析示:正常男性核型。FISH檢測示:del(17p)陽性。IGHV基因無突變。Sanger測序示TP53基因突變。患者輕度盜汗,無明顯發熱、消瘦及乏力等不適,有高血壓和糖尿病病史,無其他疾病病史和家族史。診斷為CLL(Binet A/Rai 0期)。未予以任何治療,每2~6個月隨訪1次。不是所有CLL都需要治療,只有具備至少1項治療指征時方可開始治療[4],因為過早的治療不僅不能改善患者的預後,反而可能帶來治療相關不良反應。隨著常規健康體檢的普及,這部分確診後無需立即治療的CLL患者的比例會逐漸升高。不符合指征的患者,每2~6個月隨訪1次,隨訪內容包括臨床癥狀及體征,肝、脾、淋巴結腫大情況和血常規等。該患者因為體檢發現外周血淋巴細胞比例明顯升高,通過實驗室檢查發現為成熟B淋巴細胞,流式細胞術檢測示免疫表型為典型CLL表型,診斷為CLL(Binet A/Rai 0期)。2016年發布的CLL國際預後指數(CLL-IPI)根據5個因素對預後的影響,TP53基因異常(缺失或突變)、IGHV基因無突變、β2-MG>3.5 mg/L、Binet B/C或Rai Ⅰ~Ⅳ期及年齡>5歲分別積4、2、2、1及1分,根據積分分為低危(0~1分)、中危(2~3分)、高危(4~6分)和極高危(7~10分)四個危險組,此患者積7分屬極高危組。目前暫無治療指征,建議患者每2~6個月門診隨訪,主要關注血常規、體格檢查和患者主訴。
在開始治療前,必須回答的一個問題是患者的治療指征是什麼?問題似乎很簡單,但即使在CLL常見的歐美頂級癌症中心,不同亞專科大夫對治療指征的把握也存在很大差異。美國梅奧診所血液科43位臨床醫師分成6組(其中一組為CLL專治組,余為非專治組),發現Rai 0~Ⅰ期患者從診斷至開始治療的時間:CLL專治組患者為9.2年,顯著長於非專治組的6.1年(P<0.001);不同臨床醫師治療患者的生存也有顯著差異。
在臨床實踐中我們特別注意以下幾點:①進行性血細胞減少,輕度貧血、血小板減少不必急於治療,可適當縮短隨訪期;②進行性脾、淋巴結腫大,除非有局部癥狀、脾肋緣下>6 cm或淋巴結(包括融合)直徑>10 cm才開始治療;③外周血淋巴細胞增加時間從淋巴細胞>30×109/L開始計算,需注意的是溶血、外傷、激素等可能短時間內導致淋巴細胞快速增高,一般不單純根據淋巴細胞計數開始治療;④盜汗的程度,比較嚴重的盜汗(drenching night sweat,濕透性盜汗)才是治療指征,本例患者輕度盜汗未達治療指征。另外,至今為止,早期乾預不能改善患者的生存(即使是高危患者),故本例患者儘管是極高危組也不予治療。所以,對於沒有治療指征的患者可以不進行TP53基因缺失/突變、IGHV基因突變狀態等生物學預後標誌檢測。關於CLL一線治療
例3,女,63歲。5年前確診為CLL,5年來定期隨訪疾病穩定,近6個月出現無明顯誘因體重下降10 kg(由64 kg降為54 kg),同時血常規顯示血紅蛋白和血小板進行性下降。最近一次血常規:WBC 65×109/L,淋巴細胞0.91,HGB 83 g/L,PLT 66×109/L。骨髓和外周血細胞形態學檢查示:大量成熟淋巴細胞聚集,無明顯大細胞和幼稚淋巴細胞。流式細胞術檢測示:單克隆B細胞,CD5+、CD19+、CD23+、CD200+、CD20dim、CD22-、FMC-7-。IGHV基因突變陽性,未檢測到TP53基因突變。FISH檢測示del(13q),無del(17p)、del(11q)及+12。臨床有乏力表現,近期體重下降明顯,無其他伴發疾病病史。
CLL屬於惰性白血病,但是總體而言患者病情呈現緩慢進展,大多數患者在診斷3~5年會出現治療指征,建議開始針對CLL進行治療。近年來CLL治療技術發展較快,一線治療選擇較多。一般而言,如果患者年齡<70歲特別是<65歲,IGHV基因突變陽性,無高危分子生物學和細胞遺傳學標誌,並且身體一般狀況較好(CIRS評分<6分,肌酐清除率>70 ml/min),可以採用標準的FCR方案進行治療。但是FCR方案對於老年患者治療相關不良反應尤其是感染較為常見,因此需要做好支持治療,以降低治療相關不良反應(甚至死亡)的發生率。在年齡>65歲的患者中FCR方案的不良反應增加,而療效增加不顯著。因此,對年齡>65歲、無del(17p)/TP53基因突變的體能尚好的CLL患者,國外推薦BR(苯達莫司汀聯合利妥昔單抗)方案。由於苯達莫司汀尚未在國內上市,我們採用利妥昔單抗聯合減低劑量的FC方案,也能取得滿意療效。對於年齡較大(≥70歲)和(或)伴發疾病較多(CIRS評分≥6分,肌酐清除率≥30~50 ml/min)不能耐受FCR方案,或者具有不良預後分子標誌[IGHV基因無突變、del(11q)等]的CLL患者中建議採用利妥昔單抗聯合口服苯丁酸氮芥或者單葯伊布替尼的治療方案,在能夠獲得較好治療效果的同時有效降低治療相關不良反應的發生率。本例患者系相對年齡較輕,無明顯伴發疾病,診斷5年後出現明確治療指征,並且患者無明顯不良預後標誌,因此我們採用標準FCR方案治療。經過治療患者獲得CR,目前常規隨訪中。
另外,需了解貧血的原因,此患者Ret、膽紅素、LDH均正常,Coombs試驗陰性,可以排除自身免疫性溶血性貧血(AIHA)。如存在AIHA,忌用氟達拉濱單葯治療,因其本身就可能引起致命的溶血,如採用伊布替尼也需先控制溶血。
關於高危和複發難治性CLL治療
例4,男,69歲,10年前確診為CLL,2年前患者經過FCR方案治療後獲得CR,定期隨訪疾病穩定,近3個月無明顯誘因出現低熱伴淺表淋巴結進行性腫大,同時血常規顯示血紅蛋白和血小板進行性下降,最近一次血常規:WBC 53×109/L,淋巴細胞0.89,HGB 73 g/L,PLT 76×109/L,骨髓和外周血細胞形態學檢查示大量成熟淋巴細胞聚集,幼稚淋巴細胞比例為0.040。流式細胞術檢查示:單克隆B細胞,CD5+、CD19+、CD23+、CD200+、CD20dim、CD22-、FMC-7-。IGHV基因無突變,TP53基因突變陽性。FISH檢測示del(17p)。臨床有低熱、出汗增多,患者有高血壓和輕度腎功能不全病史,無其他伴發疾病病史。
CLL目前屬於不能治癒的疾病,隨著病程的發展和多次治療,疾病會由惰性向侵襲性過程發展,因此對於高危[del(17p)和(或)TP53基因突變、對聯合免疫化療和氟達拉濱耐葯]或複發難治性CLL患者的管理目前仍然是臨床難點。總體而言,對於此類患者單純使用化療藥物大多數緩解時間較短,疾病容易出現快速進展,推薦使用新葯或新的治療模式。目前有較多臨床證據的新葯是BTK抑製劑伊布替尼、PI3K抑製劑Idealalisib+利妥昔單抗和Bcl-2抑製劑Venetoclax,雖然這些新葯不能完全克服高危和複發難治性CLL患者的不良預後,但是相較於傳統化療藥物療效顯著提高,且治療相關不良反應可控。同時對於部分相對年輕、身體情況較好的高危CLL患者可以考慮進行減低預處理強度的allo-HSCT進行鞏固治療。CAR-T細胞治療在複發難治性CLL已有報導,初步結果令人鼓舞,但是由於病例數較少,長期療效和治療風險尚需更多臨床試驗來驗證。對於本例患者,我們在確認患者疾病進展並具有治療指征後,對患者的腫瘤生物學特徵進行重新全面評估。首先淋巴結活檢仍然提示為CLL,未見大細胞轉化,但是分子生物學和細胞遺傳學檢查示del(17p)和TP53基因突變陽性,屬於高危CLL,患者既往採用過FCR方案,但是進展較快。經過綜合考慮,我們推薦該患者使用單葯伊布替尼(420 mg/d)治療,如果患者療效和身體狀況較好,可以在有合適供者的前提下進行減低預處理強度的allo-HSCT或參加臨床試驗。如HBsAg及HBV DNA陽性,服用伊布替尼的同時需抗HBV治療,預防HBV再激活。此例患者為HBsAg及HBV DNA陰性。
總結
臨床工作中,規範化診斷、規範化治療以及根據預後因素分層治療對CLL患者預後非常重要。觀察和等待仍是早期、無癥狀CLL患者的標準治療;對於體能狀態良好,且無高危分子生物學和細胞遺傳學標誌的患者,FCR方案依舊是標準治療方案;新葯的出現為高齡、體弱患者帶來了治癒的希望,而這些新葯在del(17p)/TP53基因突變陽性和複發難治性CLL患者中也展示出令人鼓舞的結果,但這還需要進一步研究去證實。另外,值得強調的是支持治療在CLL的治療過程中具有重要作用,即使伊布替尼等新葯不良反應較少,但感染等也應引起足夠重視,疫苗接種預防感染值得進一步探討。