本文來源:中華醫學雜誌, 2018,98(24) : 1965-1967.
病歷摘要
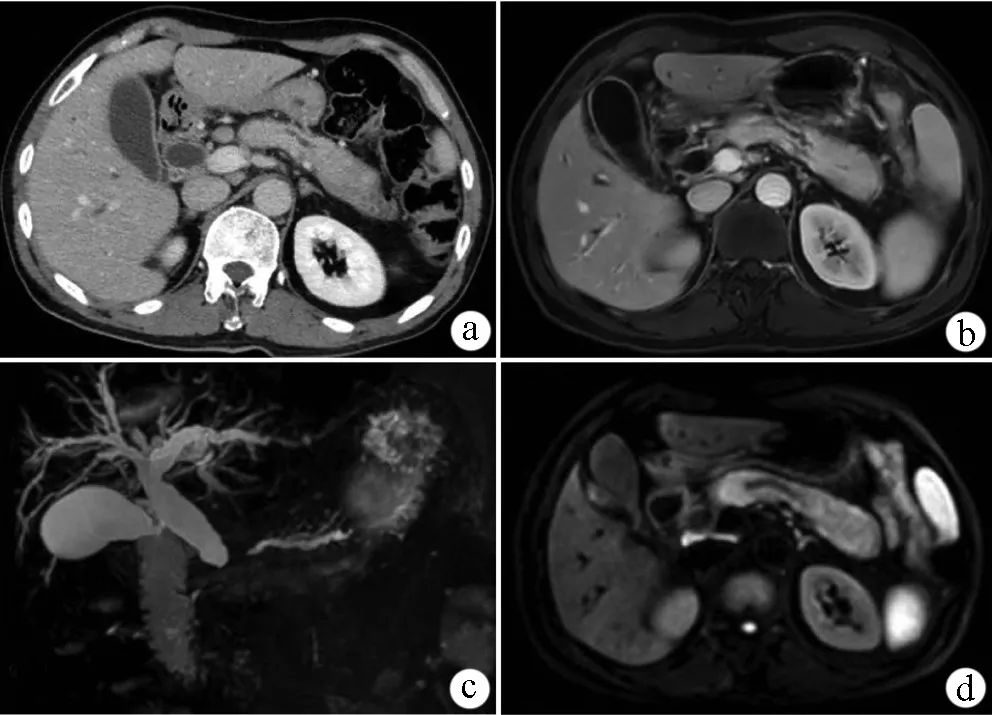
患者女,56歲,因怕冷10年、多尿6年於2017年7月19日入住北京協和醫院。患者自2007年起無誘因出現怕冷,伴顏面、手足水腫,乏力。無發熱、尿量改變。2008年9月查促甲狀腺激素(TSH)5.55 mU/L(參考範圍:0.38~4.34 mU/L),遊離三碘甲狀腺原氨酸(FT3)4.70 pmol/L(參考範圍:2.76~6.30 pmol/L),遊離甲狀腺素(FT4)16.9 pmol/L(參考範圍:10.5~24.4 pmol/L);24 h尿遊離皮質醇(24 h UFC)正常範圍,未特殊治療。2011年起無誘因出現多尿、煩渴、多飲,逐漸加重,每日飲水量及尿量約7 L,夜間尿量約4 L,夜尿5~6次。查血糖正常;TSH 10.4 mU/L,FT34正常範圍,甲狀腺過氧化物酶抗體(TPOAb)、甲狀腺球蛋白抗體(TgAb)陽性;甲狀腺超聲示甲狀腺體積增大、其內回聲粗糙呈網格狀、血流信號豐富。給予左甲狀腺素鈉口服,由12.5 μg/次,1次/d逐漸加量至100 μg/次,1次/d,複查TSH降至正常。2017年7月查尿比重1.005;血鈉147 mmol/L(參考範圍:135~145 mmol/L);紅細胞沉降率22 mm/1 h(參考範圍:0~20 mm/1 h);鞍區增強磁共振成像(MRI)示垂體柄增粗、垂體後葉短T1信號消失(圖1);行禁水試驗結果符合尿崩症;全身骨顯像示左側顱骨頂部異常放射性濃聚影。給予彌凝(去氨加壓素)口服0.05 mg/次,2次/d,患者尿量減少至2~2.5 L/d,夜尿1次。為進一步診治入院。既往史、個人史、婚育史無殊,49歲絕經。家族史:兩個哥哥分別患有自身免疫性肝炎、腎炎,一個姐姐患有系統性紅斑狼瘡。

註:A.治療前垂體柄增粗病變(↑);B.治療1個月後病變明顯縮小(↑)
▲圖1 治療前後患者鞍區MRI表現
入院查體:生命體征平穩;口唇黏膜乾燥,未見皮疹;淺表淋巴結無腫大;甲狀腺無腫大,質硬;雙乳Ⅴ期,無觸發泌乳;陰毛Ⅲ期;心、肺、腹查體無殊;眼瞼及雙下肢無水腫。血常規、糞便常規、肝腎功能、血糖、凝血功能正常;血滲透壓305 mOsm/kg·H222O;甲狀腺功能正常,TPOAb 173 U/L(參考範圍:<34 U/L),TgAb>4 000 U/L(參考範圍:<115 U/L);垂體前葉功能正常;β人絨毛膜促性腺激素(β-HCG)、甲胎蛋白(AFP)等腫瘤標誌物正常;紅細胞沉降率38 mm/1 h,超敏C反應蛋白4.91 mg/L(參考範圍:0~3.00 mg/L),類風濕因子42 U/L(參考範圍:0~20 U/L);免疫球蛋白G(IgG)17.45 g/L(參考範圍:7.00~17.00 g/L),IgG4為5 650 mg/L(參考範圍:80~1 400 mg/L),總IgE 316 kU/L(參考範圍:0~60 kU/L),IgA、IgM、IgG1/2/3、補體正常範圍;抗核抗體為核仁型1∶80陽性;腦脊液壓力、常規、生化、β-HCG、AFP正常;淚腺、涎腺、頸部淋巴結超聲正常;磁共振胰腺膽管成像未見胰膽管異常,見左腎部分實質異常信號,完善增強CT提示左腎部分實質灌注異常、缺血纖維化改變可能性大(圖2);頭顱CT示枕骨局部內板及板障缺如,符合侵蝕破壞(圖3);胸部高分辨CT示右肺中葉斑片影及索條影,雙肺胸膜下少許磨玻璃影。完善超聲引導下甲狀腺穿刺活檢術,超聲見甲狀腺瀰漫性病變,病理示濾泡萎縮,可見多量淋巴細胞、漿細胞浸潤(圖4),結合免疫組化結果:AE1/AE3(局灶+),Ki-67(index 5%),TTF-1(局灶+), CD3(+),CD20(+),CD38(+),CD138 (+),CD1a(-),S-100(-),CD68(+),IgG(+),IgG4(+),IgG4/IgG>40%,符合IgG4相關性纖維性甲狀腺炎。考慮IgG4相關性疾病(IgG4-related disease,IgG4-RD)診斷明確,加用潑尼鬆口服40 mg/次,1次/d;嗎替麥考酚酯口服0.75 g/次,2次/d,繼續彌凝、左甲狀腺素鈉治療,4個月後潑尼松已減量至20 mg/次,1次/d,嗎替麥考酚酯減量至0.5 g/次,2次/d,左甲狀腺素鈉減量至75 μg/次,1次/d,複查IgG4為853 mg/L,紅細胞沉降率、超敏C反應蛋白正常,複查鞍區MRI示垂體柄病變明顯縮小(圖1)。
▲圖2 患者腎臟增強CT示左腎部分實質灌注異常(↑)
▲圖3 患者頭顱CT示枕骨局部內板及板障缺如(↑)
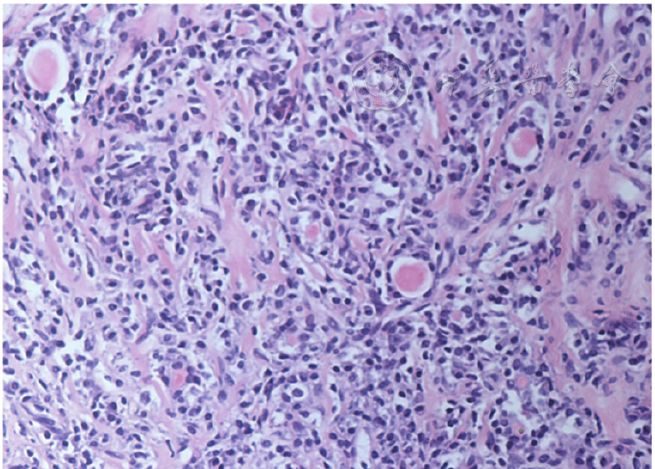
▲圖4 甲狀腺組織病理結果符合纖維性甲狀腺炎(HE ×100)
診斷難點
IgG4-RD屬於罕見疾病,除了淋巴結、涎腺、淚腺、胰腺等常見受累部位,亦可累及垂體、甲狀腺、骨骼,對於多系統受累的患者,應注意篩查IgG4-RD等自身免疫性疾病。
血清IgG4水準升高亦可見於非IgG4-RD,診斷IgG4-RD需要除外惡性腫瘤、結締組織病等,因此獲取組織病理診斷尤為重要。
啟示
垂體柄活檢風險較高,對於同時存在其他臟器受累的患者,可選取較為表淺的臟器進行組織活檢。
-
IgG4-RD對糖皮質激素治療反應良好,糖皮質激素治療後垂體柄增粗病變縮小,可進一步驗證IgG4相關性垂體炎的診斷。
分析與討論
患者中年女性,慢性病程,起病隱匿。主因甲狀腺功能減退、多尿就診,結合查體及輔助檢查結果,考慮慢性甲狀腺炎、中樞性尿崩症診斷基本明確,同時存在顱骨侵蝕性病變。鑒於患者同時存在甲狀腺、垂體及骨骼受累,從一元論的角度,入院時鑒別診斷主要考慮:(1)朗格漢斯細胞組織細胞增生症(LCH),常累及骨骼、皮膚、淋巴結、肺、肝臟、脾臟、骨髓、中樞神經系統、甲狀腺等器官,但明確診斷需要獲取組織病理;(2)自身免疫性疾病,包括IgG4-RD等,支持點包括可以引起多系統受累、患者存在自身免疫性疾病家族史、紅細胞沉降率輕度增快。入院後完善相關檢查後,血清IgG4水準明顯升高伴總IgE升高支持IgG4-RD,但IgG4-RD常見受累器官(包括淋巴結、涎腺、淚腺、胰腺、膽管及腹膜後間隙)相關篩查均為陰性,且垂體、甲狀腺均為IgG4-RD少見受累器官,骨骼受累更為罕見,此為不支持點。LCH方面,雖然LCH可以解釋垂體、甲狀腺、骨骼的病變,但LCH很少累及腎臟,無法解釋腎實質缺血纖維化性改變。此時明確診斷的關鍵在於獲取組織病理證據。經過權衡組織活檢的創傷性、風險及診斷意義,考慮垂體及顱骨(病灶臨近靜脈竇)活檢風險較高,最終選擇對甲狀腺組織進行活檢,但超聲示甲狀腺為瀰漫性病變,無局灶性結節樣病灶可以指導活檢部位的選取,因此活檢後是否能夠獲得有意義的病理結果亦未可知。通過與患者及家屬充分溝通,行超聲引導下甲狀腺粗針穿刺活檢術,病理示甲狀腺組織濾泡萎縮,可見多量淋巴細胞、漿細胞浸潤,IgG4陽性漿細胞/IgG陽性漿細胞>40%,符合IgG4相關性纖維性甲狀腺炎,同時CD1a、S-100免疫組化染色陰性,不支持LCH診斷。根據2011年IgG4-RD綜合診斷標準,患者存在多個臟器腫大、血清IgG4≥1 350 mg/L、組織病理學符合IgG4-RD,且無惡性腫瘤、結締組織病等疾病證據,可確診IgG4-RD。IgG4-RD的好發年齡為54~67歲,本例患者處於好發年齡階段內。
甲狀腺、垂體均為IgG4-RD的少見受累器官,根據北京協和醫院對346例IgG4-RD器官累及情況分析,甲狀腺、垂體受累的發生率分別為4.0%、2.3%。目前尚無IgG4相關性甲狀腺炎的器官特異性診斷標準。IgG4-RD累及甲狀腺的臨床表現缺乏特異性,常表現為甲狀腺單側或雙側腫大,質地堅韌,可伴有局部壓迫癥狀、甲狀腺功能減退相關癥狀。病理表現方面,大體觀與慢性淋巴細胞性甲狀腺炎(又稱橋本甲狀腺炎)纖維化亞型或纖維性甲狀腺炎(又稱Riedel甲狀腺炎)相似,鏡下可見甲狀腺濾泡萎縮,顯著纖維化。密集淋巴細胞及漿細胞浸潤、席紋狀纖維化、閉塞性靜脈炎是3個主要的病理學特點(其中閉塞性靜脈炎在甲狀腺組織中相對少見),此外,IgG4陽性漿細胞計數與IgG4陽性漿細胞/IgG陽性漿細胞的比例是診斷必備要素,也是與非IgG4相關性橋本甲狀腺炎、Riedel甲狀腺炎鑒別的要點。本例患者可診斷IgG4相關性Riedel甲狀腺炎。
IgG4相關性垂體炎的診斷標準於2011年由Leporati等提出,包括:(1)垂體組織病理學示垂體單核細胞浸潤,富含淋巴細胞和漿細胞,每個高倍鏡視野IgG4陽性細胞>10個;(2)垂體MRI示鞍區腫塊或垂體柄增粗;(3)活檢證明其他器官存在IgG4相關性病變;(4)血清IgG4升高,≥1 400 mg/L;(5)使用糖皮質激素後垂體腫塊變小,癥狀改善。當滿足以下3項之一時,診斷成立:標準(1),或標準(2)+(3),或標準(2)+(4)+(5)。Shikuma等總結了84例符合Leporati診斷標準的IgG4相關性垂體炎病例,26.2%的病例表現為垂體前葉功能減退,17.9%表現為中樞性尿崩症,52.4%表現為全垂體功能減退。合併受累器官中,最常見的為腹膜後間隙(26.2%),其次為唾液腺(25.0%)、淋巴結(23.8%)、肺臟(20.2%)等,僅1例(佔1.2%)合併甲狀腺病變,無病例合併骨病變。本患者符合Leporati診斷標準的(2)+(3)+(4),考慮IgG4相關性垂體炎診斷明確,且加用糖皮質激素及免疫抑製劑治療後垂體柄增粗較前明顯縮小,進一步支持該診斷。
IgG4-RD中骨骼受累極為罕見,目前國際上僅有4例顳骨受累的病例報導,其中1例為雙側病變、3例為單側病變,主要累及中耳、乳突,可有骨質破壞。4例患者因為出現了嚴重的併發症(3例存在聽力下降,1例存在右乙狀竇閉塞繼發顱壓升高),均接受了手術治療,術後給予激素(單用或聯合免疫抑製劑)治療,病情控制良好。本例患者骨骼病變因為部位臨近靜脈竇,且無明顯臨床癥狀,未行病灶活檢或手術乾預,目前無法確證是否為IgG4-RD相關,暫以一元論解釋,日後可隨訪該病灶變化是否與其他病灶保持一致。
IgG4相關性腎臟病(IgG4-RKD)並不少見,根據其診斷標準,本例患者存在異常腎臟影像學表現(增強CT中的多發低密度病灶)、血清IgG4水準升高、其他器官的組織病理診斷符合IgG4-RD,可確診IgG4-RKD。雖然目前尚無尿檢異常或腎功能下降表現,日後須密切監測腎臟相關指標。
治療方面,本患者IgG4-RD診斷明確,存在多種重要臟器受累,有積極治療指征。根據2015年IgG4-RD管理和治療的國際指南共識,糖皮質激素是治療首選藥物,推薦劑量為30~40 mg/d,可根據體重和病情進行適當調整。多數患者病情改善。當病情得到控制後應規律減量,至最小維持量維持。應注意的是,停用激素病情複發率高,部分患者即使使用小劑量激素維持,病情也可能複發,因此治療過程中應密切監測病情變化。推薦免疫抑製劑可在以下情況下使用:(1)病情活動而不能遞減激素時應該聯合免疫抑製劑;(2)小劑量激素維持時病情複發者;(3)部分專家建議在多器官受累,病情較重的患者起始治療時也可加用免疫抑製劑聯合治療。常用的免疫抑製劑包括硫唑嘌呤、嗎替麥考酚酯、甲氨蝶呤、他克莫司、環磷醯胺等。