不知道你能想像的葯最貴是多少錢?
1萬,10萬,100萬?......
近日,經美國FDA批準
小兒脊髓性肌肉萎縮基因療法在美上市
一支藥物標價210萬美元(約1448萬元)
堪稱史上最貴葯
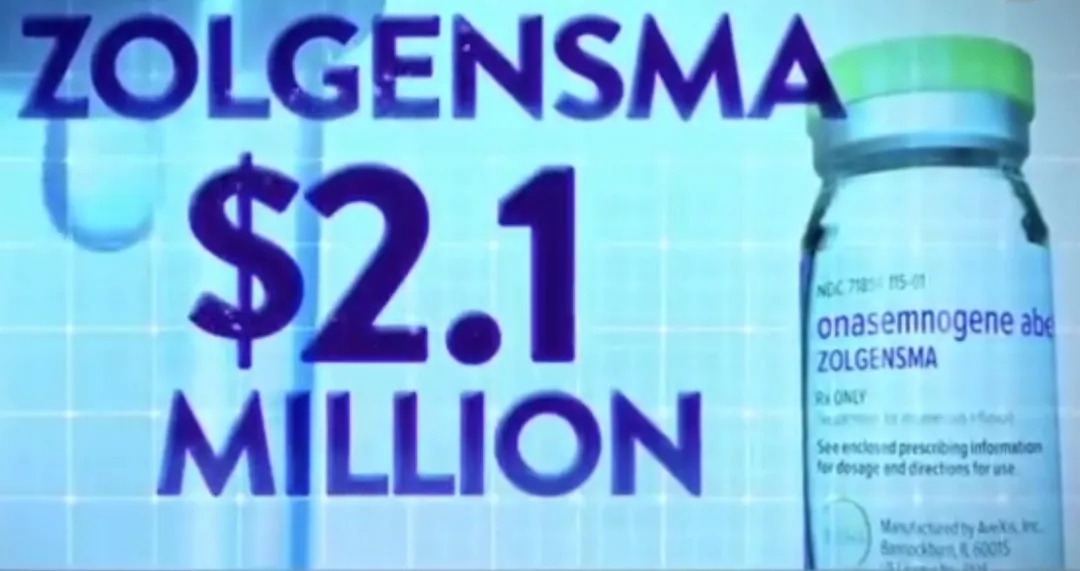

為什麼這款葯這麼貴
卻還敢稱公道價?
首先要從這個葯治療的疾病SMA說起
什麼是SMA疾病?
脊髓性肌萎縮症(spinal muscular atrophy,SMA)是一種由脊髓前角運動神經元退化引起的神經肌肉性疾病,患者主要表現為進行性、對稱性四肢和軀乾肌肉無力、萎縮,重症患兒常死於呼吸衰竭。
SMA是造成嬰幼兒死亡最常見的常染色體隱性遺傳病之一,不同人群中的致病基因攜帶頻率在1/40~1/50,新生兒的發病率約為1/10000。如果按照中國每年1700萬左右的出生人口計算,就意味著SMA每年可能會給1700個家庭帶來苦難。


也有人會問,這感覺跟霍金的病有點兒像呀!
不一樣的,霍金的病叫做肌萎縮側索硬化(ALS),也叫作運動神經元病,也就是我們所熟知的「漸凍症」,是一種漸進性的神經退行性疾病,發病年齡往往在40-50歲;而SMA是一種遺傳病,大多數情況下一出生就會發病。當然,這二者都屬於罕見病的範疇!


SMA疾病是什麼原理?
1991年,國際脊髓性肌萎縮症聯盟正式確立了SMA的分類方案,他們根據患者發病年齡和運動功能,將SMA分為Ⅰ、Ⅱ、Ⅲ 3個型別。後來,又增加了Ⅳ型和0型。
其中,Ⅰ型SMA又叫做Werdnig-Hoffman病,是最常見的類型。患兒由於嚴重肌張力減退及對稱性肌無力導致無法支撐自己的頭部,並且由於舌肌和咽肌萎縮,患兒會出現餵養和呼吸困難,通常會因為呼吸衰竭而活不過2歲。
Ⅱ型SMA被稱為中間型SMA。患兒常伴有關節攣縮和脊柱側凸,隨著病情的進展需要呼吸機維持呼吸,可存活至2歲以後。

 播放GIF
播放GIF
Ⅲ型SMA也被稱為Kugelberg-Welander病,患者的生存基本不受影響,可能在某些時刻需要輪椅。與Ⅱ型SMA不同,絕大部分Ⅲ型SMA患者的脊柱不會出現側凸,也很少有呼吸肌無力的癥狀,他們的壽命基本不受影響。


還有不到5%的SMA患者是Ⅳ型SMA,這是病症最輕微的類型,他們的癥狀類似於Ⅲ型SMA,但多是在成年後發病,一般在30歲或更晚才發病。
此外,SMA還有一種不常見的類型,發生在宮內,表現為胎動減少,患兒出生時無反射、面部癱瘓和關節攣縮,稱為0型SMA。通常這類患兒的自然死亡年齡不超過1個月……
值得一提的是,儘管SMA患者的運動功能會受到不同程度的影響,但他們的認知是正常的。
這款神奇的葯真的能治好嗎?
據報導,這款新療法名為Zolgensma,是一種基於腺相關病毒載體的基因療法,靜脈給葯可能可以修復相關基因缺陷,從而改善肌肉功能,提高患者生存率。
Zolgensma的獲批基於一項已經完成的1期臨床試驗START,以及一項正在進行中的3期臨床試驗STR1V。這兩項臨床試驗共入選36位罹患嬰兒型脊髓性肌萎縮症的小兒科患者,研究起始時這些患者年齡約在2周~8個月之間。
在已經完成的START臨床試驗中,15名患者接受了Zolgensma的治療,在接受治療後24個月的隨訪時,所有患者仍然存活,且不需要永久性的呼吸支持。


在正在進行的STR1V臨床試驗中,21名患者接受了Zolgensma的治療,截至隨訪數據截止日期,即2019年3月,19名患者依然存活,且不需要永久性的呼吸支持,1名患者死於疾病進展,1名患者退出。在19名依然存活的患者中,13名患者年齡至少有14個月大。
與自然歷史數據相比,這些臨床試驗數據證明了Zolgensma的有效性。這一疾病的自然歷史數據顯示,超過90%患者在24個月時會死亡或者需要永久性呼吸支持。
基因治療技術的發展將為無數遺傳病患者帶來了新希望。比如我院兒科遺傳代謝病實驗室利用AAV病毒結合CRISPR/Cas9基因編輯技術在治療小鼠甲基丙二酸血症和鳥氨酸氨甲醯轉移酶缺乏症模型中取得了顯著的研究成果,這為遺傳代謝病的臨床基因治療提供了重要的理論和技術支持。
言而總之,
雖然藥物很昂貴,
但生命是無價的!
中六君想,
正因為對於生命的渴求,
才有了這麼多新的技術革新...
責任編輯:郭松青
初審:李饒堯
審核:簡文楊
審定發布:李冠宏
文章部分內容來源於微博、丁香園
【感謝兒科郝虎副主任、
神經科蔡曉冬副主任對本文的支持】
圖/銳景創意,部分圖片來源於網路
TAG: |