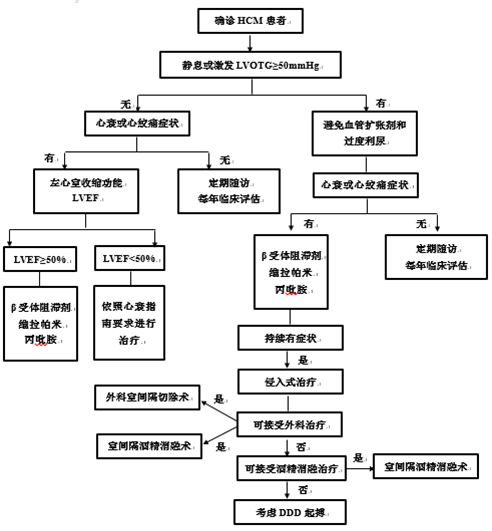
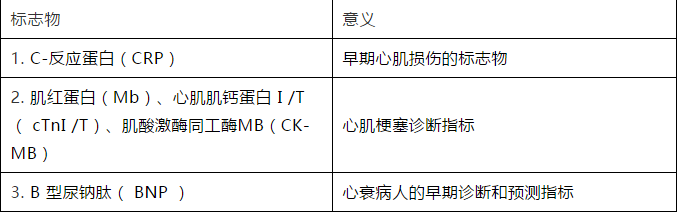
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一類最常見的單基因常染色體顯性遺傳性心肌病,也是青少年運動性猝死的最常見病因。HCM以不明原因的心室對稱性或非對稱性肥厚為主要特徵,組織病理學改變具有一定的特異性,特點為心肌細胞肥大、排列紊亂以及纖維化。編碼肌小節蛋白的基因突變是引起HCM的主要原因,即肌小節性HCM。目前已知導致HCM的突變基因有很多種,其中以MYH7,MYBPC3,TNNT2基因突變最為主要。如今HCM的診斷還主要依賴基因檢測、臨床表現以及超聲心動圖檢查。對於HCM高危患者,通過基因檢測可作出早期診斷,儘早採取相應積極乾預手段,避免嚴重病情的產生。臨床表現類似HCM的疾病有很多種,稱為HCM擬表型疾病,臨床上及時進行相應鑒別診斷對於HCM的精準治療以及預後是非常重要的。本文對幾種常見的HCM基因、臨床診斷依據以及HCM擬表型疾病的鑒別診斷的現狀做一介紹。
HCM的基因型
近幾十年關於HCM分子遺傳學的研究已經明確,HCM是一類常見的單基因常染色體顯性遺傳病,具有明顯的遺傳異質性,其不同的致病基因可能是導致HCM異質性的原因之一。目前報導已發現近40種基因,1500多個位點與HCM的發病相關,主要為編碼肌小節的結構蛋白基因。以β肌球蛋白重鏈基因(beta myosin heavy chain,MYH7)、肌球蛋白結合蛋白C基因(myosin binding protein C,MYBPC3)和肌鈣蛋白T(cardiac troponin-T,TNNT2)這三個編碼肌小節結構蛋白基因為最主要的致病基因型。前兩種是我國HCM患者的主要致病基因,其中MYH7基因主要位於染色體14q12位點,以點突變為主要突變形式。有研究認為相比於其他突變基因,MYH7基因突變幾乎完全表現為外顯且發生猝死幾率高,發病年齡更小,心肌肥厚程度更重,預後差。MYBPC3基因主要位於11p11.2位點,以截短突變為主,發病較晚且預後良好。TNNT2基因位點為1q32,以錯義、缺失突變為主,易發心臟性猝死。其他常見的突變基因及其位點可見表1所示。致病基因引發肌節蛋白的改變存在於正常肌小節中,被認為是心臟收縮功能減低的原因,並可引起肌細胞張力增加。因此,研究HCM的致病突變基因十分重要,可以用於疾病的基因篩查。基因檢測不僅特異性高,發現存在致病基因的突變即可確診,有利於儘早對高危患者採取乾預措施,同時也可作為與HCM擬表型疾病進行鑒別診斷的依據。
表1 肥厚型心肌病常見致病基因及其位點
| MYH7 | β肌球蛋白重鏈 | 14q12 | 35%~50% |
| MYBPC3 | 肌球蛋白結合蛋白C | 11p11.2 | >20% |
| TNNT2 | 肌鈣蛋白T | 1q32 | >20%(我國<2%) |
| TPM1 | 原肌球蛋白 | 15q22.1 | >5% |
| TNNI3 | 肌鈣蛋白I | 19q13.2 | >5% |
| MYL3 | 肌球蛋白必要輕鏈 | 3p21.3~p21.2 | <5% |
| MYL2 | 肌球蛋白調節輕鏈 | 12q23~q24.3 | <5% |
| ACTC | α肌動蛋白 | 11q | <5% |
| CSRP3 | 肌肉LM蛋白 | 11q15.1 | <2% |
| TTN | 巨絲蛋白 | 2q24.3 | <0.5% |
| TNNC1 | 心肌肌鈣蛋白C | 3p21.3 | 罕見 |
| MYH6 | α-肌球蛋白重鏈 | 14q | 罕見 |
| PRKAG2 | 蛋白激酶A,活動性激酶AMP β亞基 | 7q22~q31.1 | 未知 |
| MTTI | 線粒體DNA | 線粒體 | 罕見 |
HCM的臨床診斷
1.體格檢查和臨床表現:
HCM患者體格檢查可無異常發現。心前區檢查可顯示出明顯的胸骨旁心前區收縮期抬舉性搏動,頸靜脈搏動在收縮早期增強,存在左心室流出道梗阻時減弱。有流出道梗阻患者由於心瓣膜延遲關閉,聽診時會出現第二心音反常分裂。HCM患者中被證實常存在兩種雜音。第一種雜音是由於二尖瓣收縮期前向運動(SAM),導致二尖瓣前葉功能異常,二尖瓣反流引發心尖收縮中期雜音並向腋下放射。這種雜音隨著心臟收縮時相變化可能發生變化。第二種雜音是由於血液以湍流形式流經左心室流出道產生,特點為收縮中期粗糙的漸強漸弱型雜音,胸骨左緣下部雜音最響亮。減少前負荷的措施如Valsalva動作,由蹲位變站位等會引起HCM患者雜音強度增加。相反增加前負荷如站位變蹲位或者被動腿抬高等將引起HCM患者雜音強度降低。這也是HCM區別於其他疾病的特徵之一。
HCM的臨床表現具有異質性。最常見的癥狀是由於左心室肥厚(left ventricular hypertrophy,LVH)而左心室舒張末壓增高造成的呼吸困難。存在左心室流出道梗阻者心肌收縮加強而二尖瓣反流更嚴重,因而呼吸困難加劇,同時心室壁張力增加導致心肌需氧量進一步增加。心肌細胞排列紊亂和纖維化是HCM患者心室舒張障礙和心律失常的基礎,出現室性心律失常後可引發暈厥。合併流出道梗阻者,心肌收縮力增加,流出道壓力階差增大,心排血量不隨運動量增加而引發暈厥。HCM最常見的心律失常是陣發性心房顫動,最新研究表明,心房顫動並不是HCM致死性心力衰竭或致命性心律失常性猝死的原因,而心房顫動所引發的栓塞卒中則是HCM死亡的風險因素。HCM伴有陣發性心房顫動的患者經及時有效地抗凝、消融等臨床治療,可預防其血栓栓塞的發生,同時降低陣發性心房顫動轉化為持續性心房顫動的可能,預後良好。現今對於HCM伴發心房顫動患者發生血栓的風險預測,仍是需要進一步攻克的難題。需特別注意的是心室顫動等惡性心律失常所引發的心原性猝死可以是HCM的首發癥狀,可見於青年運動員劇烈運動時。
2.心電圖表現:
心電圖異常幾乎存在於所有HCM患者,其靈敏度高但特異性差。典型心電圖表現為LVH和左心房增大。常見心電圖表現以T波和ST-T改變發生率最高,大多數HCM患者ST段呈水準壓低,壓低幅度為0.1~0.5 mV;T波倒置<1 mV,主要表現在Ⅰ、Ⅱ、aVL和V4-6導聯;異常Q波也較為常見,主要存在於Ⅱ、Ⅲ和aVF導聯,形狀深且窄,不具有心肌梗死Q波的動態改變特徵,可區別於心肌梗死的病理性Q波。HCM患者還多有P波增寬等表現,提示左心房增大。心電圖表現LVH且伴有V4-6導聯明顯深的倒置T波時,特別要注意心尖部室壁瘤存在;如若V3-5有窄而深的倒置T波時,需要排除心尖肥厚型心肌病,且應與Wellens綜合征區別。3.影像學檢查:
以超聲心動圖和心血管磁共振成像(MRI)為最主要診斷依據。在缺乏明確病因的條件下,舒張期室間隔厚度>15 cm或者與後壁厚度之比>1.3,可以明確診斷HCM。多普勒超聲心動圖可準確測量左心室流出道壓力階差和二尖瓣反流量,以便判斷是否存在流出道梗阻。在評估非典型位置如前外側遊離壁、心尖或後隔部的HCM時,層析成像高解析度心臟MRI優於2維(2D)超聲心動圖[9],能精確顯示各心肌壁厚度以及部位,因此更具診斷價值。MRI可準確判斷對稱性或非對稱性的左心室壁肥厚、有無流出道梗阻、左房增大等,也可通過同位素釓延遲增強(late gadolinium enhancement,LGE)來判斷心肌纖維化的範圍及程度,其原理是基於LGE與膠原分布的部位有高度相關性。HCM表現為非對稱性肥厚者,易於診斷,而對稱性肥厚、心尖肥厚、心室中部肥厚者MRI及超聲心動圖的診斷十分重要。4.心內膜下心肌活檢:
過去並不推薦對HCM患者常規使用心內膜下心肌活檢。但現今心內膜下心肌活檢成為除外遺傳性疾病以及浸潤性疾病的有效手段。目前,美國心臟病學會/美國心臟協會(ACC/AHA)的指南表明,存在診斷不明確的情況下,可以給予心內膜下心肌活檢,尤其考慮到貯積症或浸潤性心肌疾病的鑒別診斷。心肌活檢的病理診斷結合基因檢查是HCM診斷和鑒別診斷的最重要的手段。由於屬有創檢查,且過程中可能存在嚴重併發症,因此心內膜活檢難以作為HCM的常規檢查手段在臨床推廣。
HCM的鑒別診斷
1.甘糖貯積症(Danon和PRKAG2疾病):
甘糖貯積症是由於體內代謝紊亂,甘糖貯積於細胞所引發的一類代謝障礙性疾病,主要累及肝臟、心臟、腎臟、以及肌肉組織,心電圖可表現為預激綜合征或傳導異常等,累及心臟肌肉組織時易引發嚴重的LVH,以Danon疾病最常見。Danon病是一種X連鎖顯性遺傳性溶酶體甘糖貯積症,以心肌肥厚、骨骼肌病變和智力障礙三聯征為主要臨床表現,多見於男性兒童和男性青少年發病,其引發的心肌肥厚病變與肌小節性DCM相似。由於編碼溶酶體相關膜蛋白Ⅱ基因(LAMP2)突變導致機體在甘糖降解途徑中存在缺陷,體內甘糖不斷積累,心肌細胞內大量甘糖堆積導致心肌細胞肥大和結構破壞從而引發DCM徵象。至今已發現超過60個突變位點是導致LAMP2的缺陷基因,包括移碼、拚接、缺失和截短突變[9]。區別於肌小節性的HCM,Danon患者血清肌酸激酶水準升高,大多數的患者心電圖表現為短PR間期和心室預激表現,骨骼肌活檢顯示肌質內周期性出現酸化Schiff陽性空泡。通過基因檢測篩選出含LAMP2突變基因可作為確診Danon病的依據。但對於此類疾病,至今為止沒有明確的醫學治療手段。PRKAG2心臟綜合征是一類罕見的心臟代謝性常染色體顯性遺傳病,由於編碼單磷酸腺苷激活蛋白激酶γ2亞基的基因缺陷導致體內甘糖過量沉積引起心肌細胞肥大表現。相比於肌小節性HCM,PRKAG2心臟綜合征具有心室預激、進行性傳導障礙和心臟肥大等典型表現,早期進行性收縮期功能障礙和心臟擴大也是區別於肌小節性HCM的重要特徵。
2.溶酶體貯積症(Fabry病) :
Fabry病是一類由α-半乳糖苷酶A(α-GAL A)的基因突變引起中性糖鞘磷脂在體內各器官沉積的X連鎖遺傳性染色體疾病。該病以男性兒童和年輕男性患者最多見,疾病容易引發機體多臟器功能異常,如腎臟功能衰竭,心臟功能障礙以及腦血管栓塞,其中以累及腎臟疾病的發病率最高。臨床上對於心肌肥厚合併腎功能異常,且存在明確心肌肥厚家族史的患者應警惕為Fabry病。超聲心動圖顯示,Fabry病存在左心室向心性肥厚,可區別於肌小節性的HCM。通過α-半乳糖苷酶A基因檢測以及鞘糖脂含量測定可明確診斷,一經診斷需立即接受酶替代治療,但至今為止Fabry病仍無法治癒。
3.浸潤性心肌病(澱粉樣變性):
浸潤性心肌病以澱粉樣性心肌病為主,是由於心肌中澱粉樣蛋白物質的胞外沉積而引起。研究證明,澱粉樣變性是由於基因缺陷導致功能蛋白障礙,易引發全身多臟器病變,累及心臟病變預後很差,常產生心力衰竭和猝死等嚴重癥狀。心電圖檢查顯示,QRS低電壓,同時可伴有束支或室內傳導阻滯、房室傳導阻滯等。超聲心動圖顯示,雙側心室肥大伴有瓣膜增厚,雙側心房擴張和舒張功能障礙。採用3維(3D)應變成像技術可以與肌小節性HCM進行區分。MRI顯示,全心心內膜下同位素LGE,是心臟澱粉樣變性的特徵表現,並可作為檢測疾病嚴重程度以及預後的指標。現今心肌澱粉樣變性的治療通常採用支持治療。
4.運動員心臟綜合征:
HCM是年輕運動員劇烈運動時發生猝死的最常見病因。長期運動訓練可導致生理性心室肥大,難與非阻塞性HCM患者進行鑒別。一般運動員心臟綜合征者左心室壁厚13~15 mm,左室腔徑大小正常(45~55 mm),無左心房擴大表現,且去除刺激條件一段時間後,左心室壁厚度下降,存在心室肥厚逆轉現象。超聲心動圖通常顯示,左心室充盈正常,區別於HCM舒張功能障礙引發的心室異常充盈。運動員心臟綜合征沒有遺傳性家族史,通過基因檢測可與肌小節性HCM進行有效鑒別。心臟運動試驗、動態監測以及MRI等檢查對於其鑒別診斷均有一定價值。綜合各類檢測指標,結合詳細病史、家族史以及基因檢測能有效區分運動員心臟綜合征與肌小節性HCM。
總結
HCM是一類常見的單基因遺傳性疾病,此類疾病發病範圍較廣,沒有明確的臨床診斷依據。一些HCM擬表型疾病與肌小節性HCM臨床表現相似,常被誤診為肌小節基因突變的HCM。因此HCM突變基因的研究以及對HCM擬表型疾病的鑒別在臨床診斷中顯得尤為重要。現今遺傳性代謝病的發現日漸增多,基因檢測對於鑒別此類疾病起到重要作用。存在明確的HCM基因突變即可確診為HCM。有HCM家族史等高危人群應進行基因和定期的超聲心動圖篩查。對於疑似HCM病例,結合患者的臨床表現、家族史、常規檢查手段、以及專業的基因檢測,可儘早做出診斷以便早期進行疾病管理,避免病情加重以及不良預後。