2017年12月15日,由中國醫師協會心力衰竭專業委員會發布首個中國肥厚型心肌病(HCM)管理指南,並發表於《中華心力衰竭和心肌病雜誌》。在第二十九屆長城國際心臟病學會議上,中國醫學科學院阜外醫院樊朝美教授對該指南進行了詳細的解讀。
HCM的臨床定義
成人HCM是指不能完全用心臟負荷異常解釋的左心室壁增厚(左心室壁最大厚度≥15mm),通常是由於基因突變所導致的最常見的遺傳性心臟病。兒童HCM是指左心室厚度超過同年齡、性別或體重指數的兒童左心室厚度平均值的標準差2倍以上。
HCM的病因
HCM是由編碼心肌肌小節蛋白的基因突變導致的常染色體顯性遺傳性心臟病。 已發現至少27個編碼心肌肌小節粗、細肌絲結構蛋白及Z盤蛋白的基因與HCM的發病相關(突變位點>1500個)。MYH7基因突變和MYBPC3突變是最常見的兩種致病基因突變(佔50%~70%)。
HCM的病理生理
1.心室壁肥厚
多為不均衡的局部性增厚。其組織病理學特徵是心肌細胞肥大、心肌細胞排列紊亂。
2.心肌纖維化
主要包括間質性纖維化和修復性纖維化兩種類型。診斷心肌纖維化的金標準是組織病理學檢查,心臟磁共振是目前檢測心肌纖維化首選無創影像學方法。
3.心肌缺血
病因主要包括心肌細胞肥大導致氧供和需求失衡以及心室壁內小冠狀動脈(冠脈)結構異常引起冠脈微血管功能障礙。此外,部分患者合併冠脈肌橋以及心外膜冠脈病變。單光子發射計算機斷層(SPECT)負荷/靜息心肌灌注顯像是目前評價心肌缺血的有效方法。
4.心室舒張功能異常
心室的舒張功能主要由心室肌的主動鬆弛性和被動舒張特性所決定。多普勒超聲心動圖檢查是目前評價左心室舒張功能最常用方法。
5.左心室流出道梗阻(LVOTO)
左心室流出道壓力階差(LVOTG)≥30 mmHg。
6.二尖瓣關閉不全(反流)和二尖瓣前葉收縮期前移(SAM現象)
HCM的診斷標準
關於HCM的診斷標準,不同指南對其表述有所差別。2011年美國心臟病學學院基金會/美國心臟協會(ACCF/AHA)的HCM診斷和治療指南中,HCM的診斷需除外其他可引起心室壁增厚的生理因素、心血管疾病或全身性疾病。
2014年歐洲心臟病學會(ESC)的HCM診斷和管理指南中,除心臟負荷增加導致的心室壁肥厚外,其他生理因素、心血管疾病或全身性疾病引起的心室壁肥厚可以歸為HCM。由於心室壁肥厚的原因多種多樣,鑒別診斷涉及多種檢查,且部分患者難以確定心肌肥厚的確切病因。
本指南採納2011年ACCF/AHA的HCM診斷標準。
HCM的鑒別診斷
心室壁增厚是HCM的典型特徵,但有多種生理性和病理性因素可導致心室壁增厚,因此,在臨床診斷HCM前需除外其他心血管疾病或系統性疾病,包括運動員心臟改變、高血壓性心臟肥厚、主動脈瓣狹窄、心肌澱粉樣變性。與其他遺傳性代謝性疾病相鑒別,如Anderson-Fabry病、Danon病、Pompe病、Friedreich共濟失調、線粒體心肌病等。
HCM的臨床分型
1.根據血流動力學分型
(1)肥厚型梗阻性心肌病:指異常肥厚心肌突入左心室腔,造成血流通道阻塞,在左心室血流通道上。
(2)肥厚型非梗阻性心肌病:無論是靜息時還是激發時左心室流出道均無壓力階差出現(LVOTG<30mmHg)。
2.根據肥厚部位分型
(1)心室間隔肥厚;(2)心尖部肥厚;(3)左心室中部肥厚;(4)左心室瀰漫性肥厚;(5)雙心室肥厚。
3.根據家族史和遺傳學規律分型(利於指導遺傳學研究和遺傳諮詢)
(1)家族性HCM:發病呈家族聚集,佔60%~70%,多為常染色體顯性遺傳。
(2)散發性HCM:無家族性聚集的HCM患者稱之為散發性HCM。
4.特殊類型
(1)心尖肥厚型心肌病:發病以男性為主,心電圖的典型特徵是巨大負相T波,超聲心動圖或心臟磁共振(CMR)檢查示舒張末期左心室(LV)心尖部室壁明顯增厚(最大厚度≥15mm),LV心尖部與後壁最大厚度之比≥1.5,預後相對較好。
(2)左心室中部梗阻性肥厚型心肌病:是HCM中相對少見的類型,特徵改變是左心室中部乳頭肌與室間隔中部心肌異常肥厚,伴有左心室心尖部與基底部心腔之間收縮末期壓力階差。患者常伴有明顯且嚴重癥狀,合併心尖部室壁瘤和附壁血栓形成的比例較高,導致心力衰竭、卒中和猝死的風險較高,預後較差。
(3)終末期肥厚型心肌病:HCM患者出現嚴重左心室收縮功能障礙(通常指LVEF<50%),LV常顯著擴大、心室壁相對變薄。常合併快速性心律失常和體循環栓塞,易發生難治性心力衰竭和心源性猝死(SCD),預後差。
心臟性猝死風險評估方法
SCD是HCM的首位死因。本指南推薦兩種SCD風險評估方法。
1.方法一
評估有無傳統SCD危險因素:(1)SCD家族史;(2)不能解釋的暈厥;(3)非持續性室性心動過速;(4)運動時血壓異常反應;(5)最大左心室壁厚度≥30mm。
2.方法二
應用SCD風險預測模型評估SCD風險。已有研究顯示,評估傳統SCD危險因素具有較高的靈敏度但特異性較差,而SCD模型具有較高的特異性但靈敏度稍差。
本指南推薦利用HCM Risk-SCD模型計算5年內發生SCD概率,評估其風險,以決定是否植入ICD進行一級預防。
HCM的治療
目前的HCM診斷和治療指南幾乎沒有相應的隨機對照臨床研究。本指南中的大部分推薦均基於觀察性隊列研究和專家共識,但仍為臨床醫生提供了各年齡段患者的治療方法。治療目標是緩解臨床癥狀,改善心臟功能,延緩疾病進展,減少死亡。所有HCM患者均應做SCD風險評估和危險分層,進行相應的預防和治療。
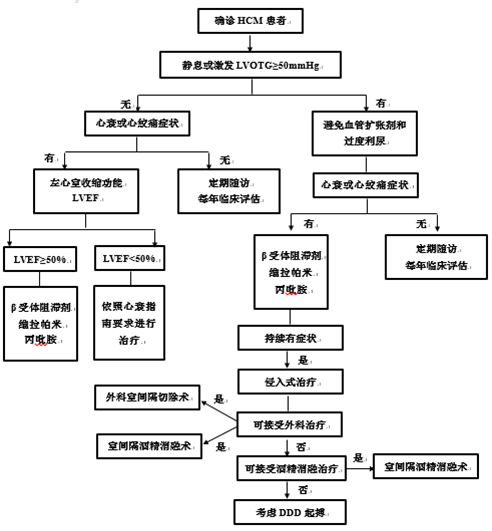
肥厚型心肌病患者治療流程圖
1.有癥狀且存在LVOTO患者
治療目標是使用藥物、外科室間隔切除術、酒精消融術或起搏器治療改善癥狀。
(1)外科室間隔切除術指征:左心室流出道峰值壓差≥50mmHg伴中重度癥狀(NYHA心功能分級III-IV級),且藥物難以控制;由於左室流出道梗阻(左室流出道峰值壓差≥50mmHg)導致反覆活動後暈厥,且藥物難以控制;同時存在其他疾病需行手術(如二尖瓣修補/置換術)。
(2)經皮室間隔心肌消融術指征:左心室流出道峰值壓差≥50mmHg伴中重度癥狀(NYHA心功能分級III-IV級),且藥物治療難以控制;由於左室流出道梗阻(左室流出道峰值壓差≥50mmHg)導致反覆活動後暈厥,且藥物治療難以控制。年齡<40歲者不推薦室間隔酒精消融術(Ⅲ類,C級);室間隔顯著增厚≥30mm患者療效不肯定,通常不考慮(Ⅱb類,C級) 。
2.有癥狀但無LVOTO患者
治療重點是管理心力衰竭、心律失常和心絞痛;對於藥物治療無效的進展性左心室收縮或舒張功能不全的患者,應考慮行心臟移植。
3.無癥狀HCM合併可能導致其他心血管疾病患者
建議每年定期臨床評估,推薦低強度的有氧訓練作為HCM患者健康生活方式的一部分。
4.無癥狀HCM有或無LVOTO患者
β受體阻滯劑和非二氫吡啶類CCB的使用是否改善臨床預後尚不明確。無論梗阻的嚴重程度,不推薦無癥狀HCM成人和具有正常耐受力的兒童HCM患者進行酒精間隔消融、外科治療。
遺傳檢測、家系篩查
HCM在基因學方面的進展非常迅速,可以進行常規的臨床應用和家系篩選。MYH7和MYBPC3的突變佔了HCM的大多數(50%~70%)。確認先證者致病基因有助於家族成員中HCM患者的早期診斷、監測和優生優育管理。建議對其一級親屬進行基因檢測及臨床評估。管理指南指出高通量二代測序技術不僅可提高致病突變的檢出率,明確更多新發突變,發現更多非常見的肌節蛋白突變。
隨訪建議
本指南推薦對HCM患者進行終生隨訪,監測癥狀變化、心血管不良事件、左心室流出道梗阻的變化情況,左心室功能和心律失常的發生風險。
根據年齡和癥狀的嚴重程度決定對患者監測的頻率。
(1)推薦對臨床狀況穩定患者進行每1~2年一次的心電圖和超聲心動圖檢查。
(2)推薦對LAd≥45mm的竇性心律患者,每隔6~12個月,進行1次48h Holter檢查,評估無癥狀性心律失常。
(3)推薦對臨床狀況穩定患者每隔5年或癥狀進展患者每隔2~3年可考慮進行CMR檢查。
(4)推薦在經皮室間隔消融治療或手術後隨訪評估應包括1~3個月內心電圖、超聲心動圖和動態心電圖。此後6~12個月1次的隨訪。
心在線 專業平台專家打造 編輯 劉明玉┆美編 高紅果┆製版 劉明玉







