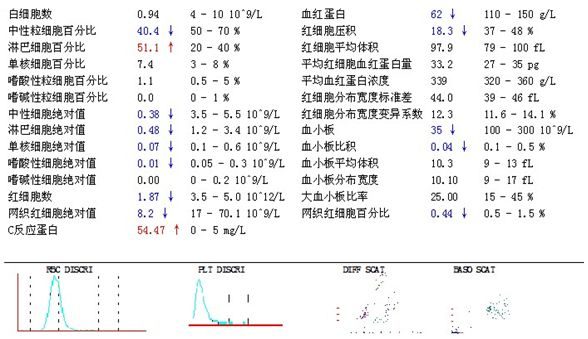
患者青壯年男性,保定市清苑區農民,主因:乏力伴皮膚紫癜2周入院。患者緣於2周前出現進食差,乏力及皮膚紫癜,3天前癥狀加重而就診於皮膚科門診,並於2017-05-5 15:40入院治療。
門診查血常規示:

WBC:272.20×10^9/L、HGB:56g/L, PLT:10×10^9 /L。外周血:白細胞總數異常增多,原始細胞佔8.0%,紅細胞大小不等,血小板較少見。(血細胞形態學如下圖示)


查體:T 37.4℃ P 90次/分 R20次/分 Bp140/80mmHg。神志清楚、精神可,貧血貌,全身皮膚可見瘀斑及散在出血點。淺表淋巴結未觸及腫大。咽無充血,扁桃體無腫大,胸骨無壓痛,雙肺呼吸音粗,未聞及明顯乾濕囉音。心音正常,心率90次/分,律齊,各瓣膜聽診區未聞及病理性雜音。腹平坦,全腹軟,無壓痛、反跳痛及肌緊張,肝未及,肋下3cm可觸及脾臟。叩鼓音,移動性濁音陰性,腸鳴音正常存在。雙下肢無水腫。
當日行骨髓象檢查(如下圖示):




骨髓增生極度活躍,原始粒細胞增多佔9.5%,以中性中幼粒細胞以下階段增生為主,可見雙核粒細胞及中性分葉粒細胞假性Pelger樣畸形。紅系增生減低,紅細胞大小不等,可見畸形紅細胞,大紅細胞,嗜鹼性點彩紅細胞,H-J小體。巨核細胞及血小板少見。骨髓細胞形態學:考慮骨髓增生異常綜合症/骨髓增殖性腫瘤(MDS/MPN),不除外不典型慢性粒細胞白血病(aCML)。


可見髓系原始細胞約佔有核細胞12.52%,並表達CD34、CD117、CD13、CD33、HLA-DR,部分表達CD56;成熟粒系比例明顯增多,考慮CML,請結合臨床除外aCML、MDS。
骨髓病理組織活檢:


骨髓活檢:骨髓增生極度活躍,粒系各階段細胞可見,偏幼稚階段細胞增多,以中幼及以下階段細胞為主,紅系細胞偏少,以中幼及以下階段細胞為主,可識別的巨核細胞偏少,分葉核為主。網狀纖維染色(MF-1級)。建議完善骨髓增生異常綜合症/骨髓增殖性腫瘤的鑒別診斷。
骨髓染色體核型分析:


染色體提示:47,XY,+8[19]/46,XY[1];克隆型異常+8。
分子生物學檢查:BCR/ABL p210、BCR/ABL p190、BCR/ABL p230均陰性。
二代測序檢查:

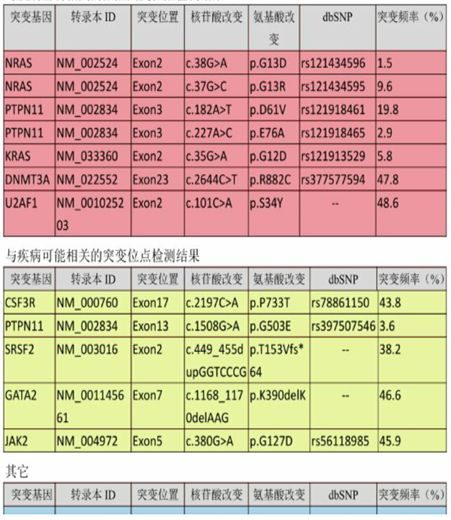
檢測到與疾病密切相關的熱點突變:NRAS、PTPN11、KRAS、DNMT3A、U2AF1。檢測到與疾病可能相關的突變點:CSF3R、PTPN11、SRSF2、GATA2、JAK2。
綜合臨床及實驗室檢查,患者最終診斷:1.骨髓增生異常綜合症/骨髓增殖性腫瘤 (MDS/MPN),2. 不典型慢性粒細胞白血病 (aCML)。給予羥基尿、鹼化、抑酸、保肝、抗感染並及時輸血、輸注血小板治療,應用地西他濱聯合小劑量阿糖胞苷、阿克拉黴素和粒細胞集落刺激因子(D-CAG)方案化療兩個療程。患者癥狀改善後暫時離院休養。期間入院兩次鞏固治療,病情相對較平穩。
2018-1-5 5:15患者急診入院,緣於發熱2天伴乏力、皮膚出血點而就醫。查體:T 37.8℃, P 90次/分,全身皮膚粘膜無黃染,雙上肢可見散在出血點,無瘀斑,周身淺表淋巴結未觸及腫大。胸骨輕壓痛,雙肺呼吸音粗,未聞及明顯乾濕性囉音。心率90次/分,律齊,腹部平坦,肝未觸及,脾大肋緣下6cm,雙下肢無水腫。神經系統查體未見明顯異常。
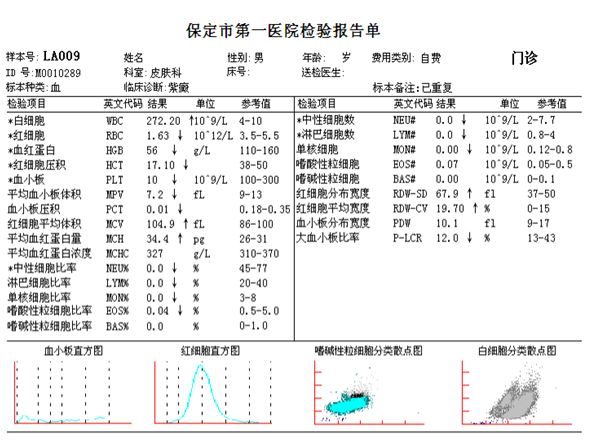
急診血常規、複查流式細胞術分別提示:


WBC:126.74×10^9/L、HGB:67.0g/L, PLT:22.0×10^9 /L。外周血:白細胞總數異常增多,原始粒細胞佔29.0%,複查骨髓流式細胞術:表型異常的髓系原始細胞佔77.86%;高度提示疾病發生演變,轉化為急性髓系白血病即AML-MRC。臨床積極對症處理,但由於患者病情危重呈進行性,且合併感染及多器官功能衰竭,轉重症醫學科治療6天后,最終而不治。
回顧:該患者初診即呈外周血高白狀態,伴明顯貧血及血小板減少,臨床表現為發熱,皮膚出血點,脾臟腫大;骨髓及外周血形態學提示:粒細胞異常增殖伴明顯發育異常,部分紅細胞伴病態造血;骨髓活檢、流式細胞術提示腫瘤異質性,BCR-ABL基因陰性,二代測序多個不良預後基因表達,遺傳學呈克隆型+8染色體改變,並綜合疾病短期即演變進展為急性髓系白血病(AML-MRC)的臨床特點,故該患者診斷: 骨髓增生異常綜合症/骨髓增殖性腫瘤(MDS/MPN)及不典型慢性粒細胞性白血病(aCML)成立。
不典型慢性粒細胞白血病( atypical chronic myelogenous leukemia, aCML)。是一種發病率低、預後差、生存期短的惡性血液系統克隆性腫瘤,費城染色體(Ph)及BCR-ABL融合基因均呈陰性,兼有骨髓發育異常和骨髓增殖的生物學特徵;在2008年WHO公布的《造血和淋巴系統腫瘤的分類標準》中,將不典型慢性粒細胞白血病(aCML)歸屬於骨髓增生異常綜合症/骨髓增殖性腫瘤(MDS/MPN)。其細胞形態學特徵為:骨髓及外周血粒系異常增殖且發育異常,伴或不伴紅系或巨核系發育異常;Ph染色體及BCR-ABL融合基因呈陰性,aCML多表達SETBP1和CSF3R基因突變,並常見NRAS和KRAS突變,部分患者還出現TET2、CBL、JAK2V617F和U2AF1突變。而1/3的aCML患者伴有+8和del(20q)。不典型慢性粒細胞白血病(aCML)預後較 Ph 染色體陽性的慢性粒細胞白血病(CML)顯著為差,大約25%—40%的aCML患者會進展為急性白血病,平均存活時間不超過20個月。國外部分學者認為造血乾細胞移植(HSCT)仍是aCML患者的首選治療方案,目前去甲基化藥物—地西他濱,在治療骨髓增生異常綜合征(MDS)及慢性粒單核細胞白血病(CMML)方面取得了較好的療效,也有應用於不典型慢性粒細胞白血病(aCML)療效相對滿意的病例報導。由於本病少見,易發生誤診,故應該引起臨床醫師及血液病實驗室診斷工作者的高度重視。
附:2016版WHO 不典型慢性粒細胞白血病(aCML)診斷標準
1.外周血白細胞增高,中性粒細胞及其前體細胞(早幼粒細胞,中幼粒細胞,晚幼粒細胞,佔白細胞比例≥10%)增多
2.粒細胞生成異常,包括染色質凝集異常,嗜鹼性粒細胞絕對數不(明顯)增多
3.嗜鹼性粒細胞比例<2%,單核細胞絕對數不(明顯)增多,單核細胞比例<10%
4.骨髓有核細胞增多,粒細胞增殖和粒系病態造血,伴或不伴有核紅細胞和巨核細胞病態造血
5.外周血和骨髓原始細胞比例<20%
6.無PDGFRA,PDGFRB,或FGFR1重排,或PCM1-JAK2融合證據
7.不符合WHO規定的CML,PMF,PV或ET診斷標準。
作者簡介:


王哲 河北省保定市第一醫院檢驗科副主任,主要從事血液病實驗室診斷工作,臨床病理檢驗專業執業醫師,臨床檢驗副主任醫師,承德醫學院內科學副教授;河北省檢驗醫學診斷學會理事,血液系統疾病檢驗診斷專業委員會常務委員兼秘書,河北省中西醫結合學會檢驗醫學專業委員會常務委員,河北省老年醫學會血液病專業委員會委員,保定市檢驗醫師協會常務委員,保定市預防醫學會衛生檢驗品質控制專業委員會常務委員,檢驗視界網特約作者。主研論文科研多篇,參編著作6部,保定市衛生系統優秀共產黨員,並榮立保定市政府記功一次。